
生物多样性 ›› 2021, Vol. 29 ›› Issue (9): 1245-1255. DOI: 10.17520/biods.2021028 cstr: 32101.14.biods.2021028
栗冬梅1,*( ), 杨卫红2, 李庆多1, 韩茜2, 宋秀平1, 潘虹2, 冯云2,*()
), 杨卫红2, 李庆多1, 韩茜2, 宋秀平1, 潘虹2, 冯云2,*()
收稿日期:2021-01-19
接受日期:2021-04-09
出版日期:2021-09-20
发布日期:2021-05-28
通讯作者:
栗冬梅,冯云
作者简介:ynfy428@163.com#共同第一作者
基金资助:
Dongmei Li1,*(), Weihong Yang2, Qingduo Li1, Xi Han2, Xiuping Song1, Hong Pan2, Yun Feng2,*()
Received:2021-01-19
Accepted:2021-04-09
Online:2021-09-20
Published:2021-05-28
Contact:
Dongmei Li,Yun Feng
About author:First author contact:#Co-first authors
摘要:
蝙蝠是很多病原微生物的自然宿主, 全球多项研究表明蝙蝠是巴尔通体(Bartonella species)的主要宿主。为了解滇西南地区蝙蝠中巴尔通体的流行特征, 我们于2015-2017年间在云南省4个地区应用网捕法捕获蝙蝠3种305只。经种类鉴定后采集肝脾组织, 提取核酸, 通过TaqMan实时荧光定量PCR方法检测巴尔通体的tmRNA基因ssrA, 并进行测序鉴定和系统发育分析。结果发现172只蝙蝠检出该基因, 总感染率为56.4%; 其中临沧、西双版纳、保山和瑞丽4个采样点的蝙蝠感染率分别为50.0% (22/44)、61.7% (29/47)、62.1% (18/29)和55.7% (103/185)。中菊头蝠(Rhinolophus affinis)、小菊头蝠(R. blythi)和棕果蝠(Rousettus leschenaultii)的感染率分别为50.0% (22/44)、62.1% (18/29)和56.9% (132/232), 差异没有统计学意义(χ2 = 1.135, P = 0.567), 表明巴尔通体在云南当地的蝙蝠种群中高度流行。定量PCR扩增产物2次扩增后测序获得37个巴尔通体ssrA序列, 属于10个系统发育分支, 其中1个为伊丽莎白巴尔通体(B. elizabethae)、特利波契巴尔通体(B. tribocorum)和克拉斯诺夫巴尔通体(B. krasnovii)的近缘种。其余序列与已知巴尔通体距离较远, 与亚洲、欧洲和美洲等其他地域来源于蝙蝠的巴尔通体近缘。遗传多样性分析显示, ssrA基因的核苷酸多样性指数(π)为0.11381 ± 0.00928, 基因型多样性指数(Hd)为0.985 ± 0.010, 形成29个基因型(单倍型), 说明云南蝙蝠巴尔通体具有丰富的遗传多样性。通过对本研究标本与全球相关序列的系统发育网络重建, 分析全球蝙蝠巴尔通体的地理和宿主分布特征, 可以看出巴尔通体与蝙蝠之间存在显著的宿主特异性关联。因此可初步确定蝙蝠-巴尔通体具有协同进化特征, 同时受到地理隔离的影响。
栗冬梅, 杨卫红, 李庆多, 韩茜, 宋秀平, 潘虹, 冯云 (2021) 巴尔通体在滇西南蝙蝠中高度流行并具有丰富的遗传变异特征. 生物多样性, 29, 1245-1255. DOI: 10.17520/biods.2021028.
Dongmei Li, Weihong Yang, Qingduo Li, Xi Han, Xiuping Song, Hong Pan, Yun Feng (2021) High prevalence and genetic variation of Bartonella species inhabiting the bats in southwestern Yunnan. Biodiversity Science, 29, 1245-1255. DOI: 10.17520/biods.2021028.
| 蝙蝠种类 Species of bats | 感染率(阳性数/检测数) Infection rate (positive/detected) | 合计 Total | |||
|---|---|---|---|---|---|
| 临沧市 Lincang City | 西双版纳州 Xishuangbanna Prefecture | 保山市 Baoshan City | 德宏州 Dehong Prefecture | ||
| 中菊头蝠 Rhinolophus affinis | 50.0% (22/44) | 0 | 0 | 0 | 50.0% (22/44) |
| 小菊头蝠 Rhinolophus blythi | 0 | 0 | 62.1% (18/29) | 0 | 62.1% (18/29) |
| 棕果蝠 Rousettus leschenaultii | 0 | 61.7% (29/47) | 0 | 55.7% (103/185) | 56.9% (132/232) |
| 合计 Total | 50.0% (22/44) | 61.7% (29/47) | 62.1% (18/29) | 55.7% (103/185) | 56.4% (172/305) |
表1 不同种类和地理来源的蝙蝠感染巴尔通体的情况
Table 1 Number of species and geographical origins of the bats infected with Bartonella spp.
| 蝙蝠种类 Species of bats | 感染率(阳性数/检测数) Infection rate (positive/detected) | 合计 Total | |||
|---|---|---|---|---|---|
| 临沧市 Lincang City | 西双版纳州 Xishuangbanna Prefecture | 保山市 Baoshan City | 德宏州 Dehong Prefecture | ||
| 中菊头蝠 Rhinolophus affinis | 50.0% (22/44) | 0 | 0 | 0 | 50.0% (22/44) |
| 小菊头蝠 Rhinolophus blythi | 0 | 0 | 62.1% (18/29) | 0 | 62.1% (18/29) |
| 棕果蝠 Rousettus leschenaultii | 0 | 61.7% (29/47) | 0 | 55.7% (103/185) | 56.9% (132/232) |
| 合计 Total | 50.0% (22/44) | 61.7% (29/47) | 62.1% (18/29) | 55.7% (103/185) | 56.4% (172/305) |
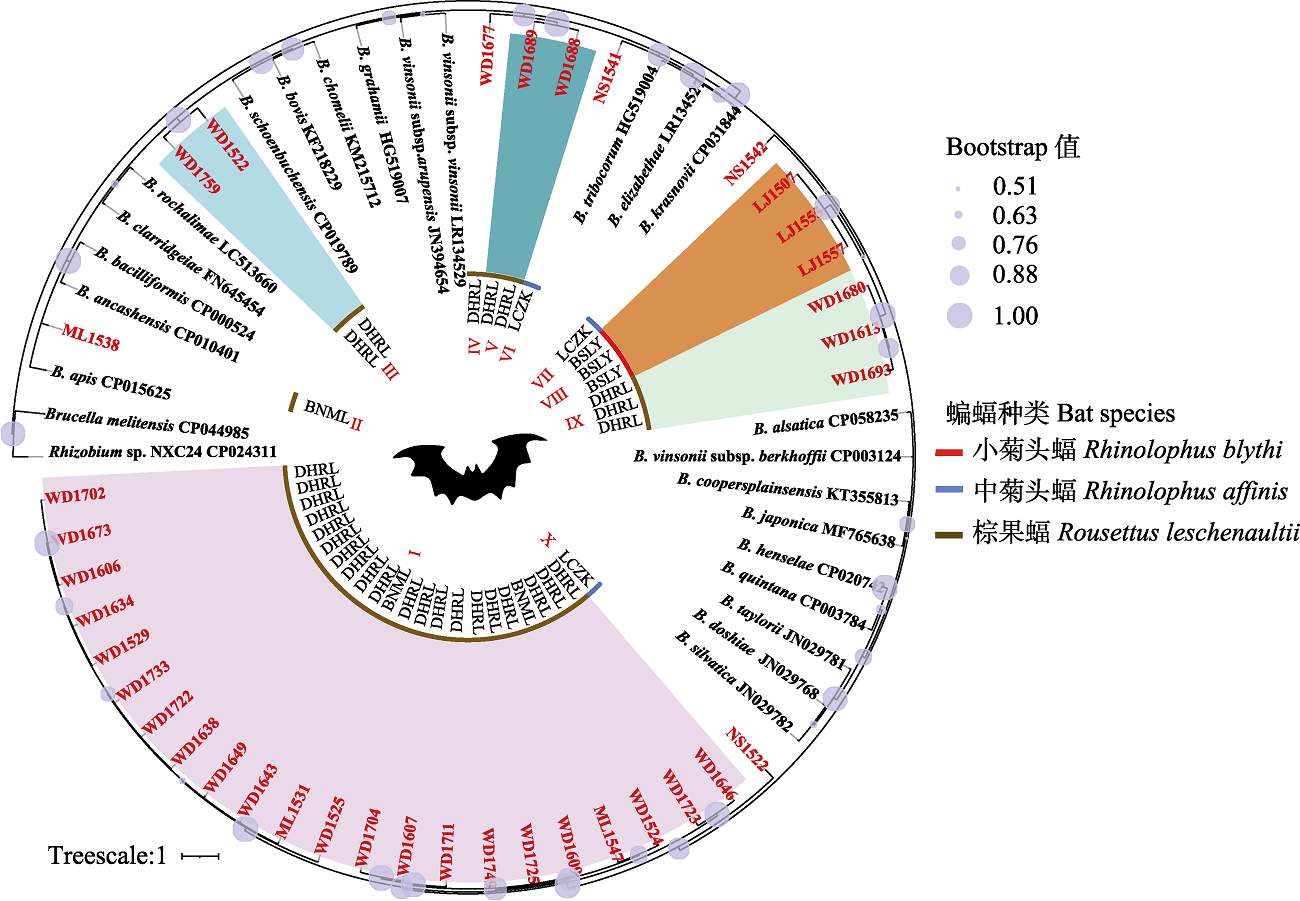
图1 基于蝙蝠标本及巴尔通体参考菌株的ssrA序列构建的贝叶斯系统发育树。蝙蝠种类在系统树中内圈, 以红、蓝和棕色线段表示; 本研究中蝙蝠序列名称用红色字体标识, 同附录3中的标本名称。图中心BSLY、LCZK、BNML和DHRL分别代表保山市隆阳区潞江镇、临沧市镇康县南伞乡、西双版纳州勐腊县瑶区乡和德宏州瑞丽市畹町镇; 图中心Ⅰ-X代表本研究中蝙蝠ssrA序列所在的进化分支; Brucella melitensis CP044985和Rhizobuim sp. NXC24 CP024311为外群。
Fig. 1 Phylogenetic tree highlighting the position of ssrA sequences of Bartonella from bats relative to reference species of Bartonella spp. The bat species in the inner circle of the phylogenetic tree, represented by red, blue, and brown lines; bat sequence names in this study are marked with red font, the same as specimen no. in Appendix 2. BSLY, Lujiang Town, Longyang district, Baoshan City; LCZK, Nansan Town, Zhenkang County, Lincang City; BNML, Yaoqu Town, Mengla County, Xishuangbanna Prefecture; DHRL, Wanding Town, Ruili City, Dehong Prefecture; I-X in the center represent the phylogroups of ssrA sequence from bats in this study, while Brucella melitensis CP044985 and Rhizobuim sp. NXC24 CP024311 are outgroups.
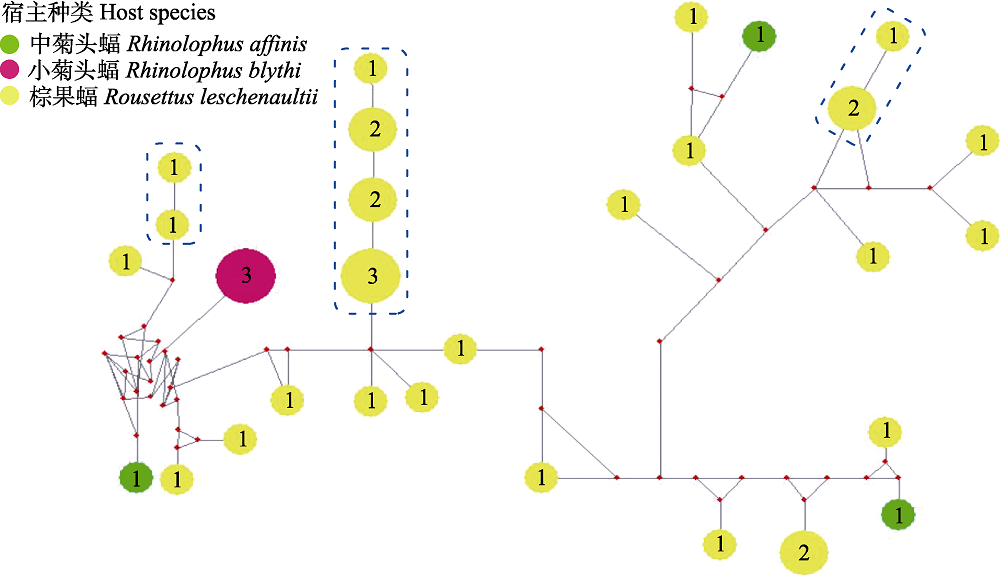
图2 基于本研究蝙蝠标本中巴尔通体ssrA序列构建的中介连接网络图。图中圆饼和其中数字分别代表基因型和个体数量, 直径与个体数量成正比。
Fig. 2 Median-joining network of ssrA genotypes for Bartonella spp. from bat specimens in this study. Genotypes are denoted as pies, in which the numbers are number of individuals, with a size proportional to genotypic frequency.
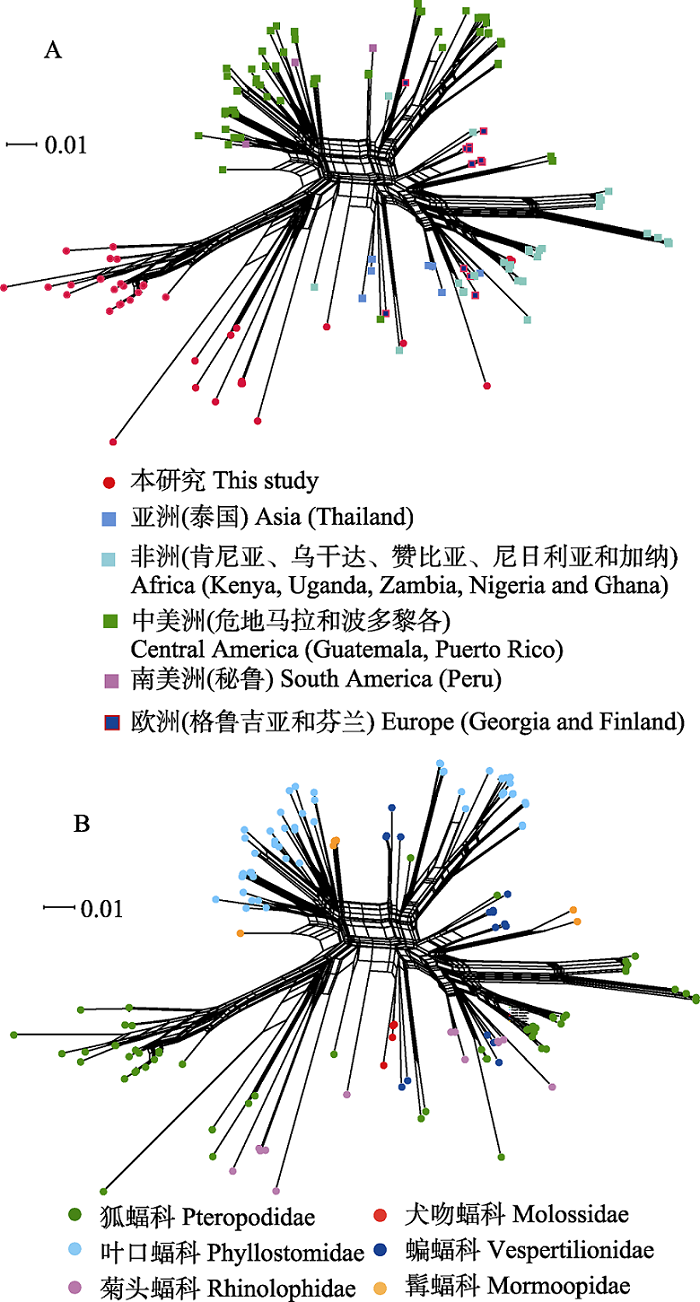
图3 基于蝙蝠标本和GenBank中巴尔通体ssrA序列构建的邻接网系统发育网络图。网络图中有152个巴尔通体ssrA序列, 其中本研究序列37个, GenBank数据库下载序列115个。 图A和B分别为依据蝙蝠宿主的地理来源和其分类绘制的网络图。
Fig. 3 Phylogenetic network diagram generated from ssrA sequences of Bartonella spp. from bat specimens in this study and GenBank using NeighborNet algorithm. There are 152 ssrA sequences of Bartonella spp. in the network diagram, including 37 sequences in this study and 115 sequences downloaded from the GenBank database. A and B are the networks of bat-borne Bartonella from the globe according to the geographic distributions and the host taxonomy associations.
| [1] |
Bai Y, Kosoy M, Recuenco S, Alvarez D, Moran D, Turmelle A, Ellison J, Garcia DL, Estevez A, Lindblade K, Rupprecht C (2011) Bartonella spp. in bats, Guatemala. Emerging Infectious Diseases, 17, 1269-1272.
DOI URL |
| [2] |
Bai Y, Osinubi MOV, Osikowicz L, McKee C, Vora NM, Rizzo MR, Recuenco S, Davis L, Niezgoda M, Ehimiyein AM, Kia GSN, Oyemakinde A, Adeniyi OS, Gbadegesin YH, Saliman OA, Ogunniyi A, Ogunkoya AB, Kosoy MY, Team IBFI (2018) Human exposure to novel Bartonella species from contact with fruit bats. Emerging Infectious Diseases, 24, 2317-2323.
DOI URL |
| [3] |
Becker DJ, Bergner LM, Bentz AB, Orton RJ, Altizer S, Streicker DG (2018) Genetic diversity, infection prevalence, and possible transmission routes of Bartonella spp. in vampire bats. PLoS Neglected Tropical Diseases, 12, e0006786.
DOI URL |
| [4] |
Concannon R, Wynn-Owen K, Simpson VR, Birtles RJ (2005) Molecular characterization of haemoparasites infecting bats (Microchiroptera) in Cornwall, UK. Parasitology, 131, 489-496.
PMID |
| [5] |
Diaz MH, Bai Y, Malania L, Winchell JM, Kosoy MY (2012) Development of a novel genus-specific real-time PCR assay for detection and differentiation of Bartonella species and genotypes. Journal of Clinical Microbiology, 50, 1645- 1649.
DOI URL |
| [6] | do Amaral RB, Lourenço EC, Famadas KM, Garcia AB, Machado RZ, André MR (2018) Molecular detection of Bartonella spp. and Rickettsia spp. in bat ectoparasites in Brazil. PLoS ONE, 13, e0198629. |
| [7] | Gutiérrez R, Vayssier-Taussat M, Buffet JP, Harrus S (2017) Guidelines for the isolation, molecular detection, and characterization of Bartonella species. Vector Borne and Zoonotic Diseases (Larchmont, NY), 17, 42-50. |
| [8] |
Han HJ, Wen HL, Zhao L, Liu JW, Luo LM, Zhou CM, Qin XR, Zhu YL, Zheng XX, Yu XJ (2017) Novel Bartonella species in insectivorous bats, Northern China. PLoS ONE, 12, e0167915.
DOI URL |
| [9] | Hornok S, Estók P, Kováts D, Flaisz B, Takács N, Szőke K, Krawczyk A, Kontschán J, Gyuranecz M, Fedák A, Farkas R, Haarsma AJ, Sprong H (2015) Screening of bat faeces for arthropod-borne apicomplexan protozoa: Babesia canis and Besnoitia besnoiti-like sequences from Chiroptera. Parasites & Vectors, 8, 1-6. |
| [10] |
Hornok S, Kovács R, Meli ML, Gönczi E, Hofmann-Lehmann R, Kontschán J, Gyuranecz M, Dán A, Molnár V (2012) First detection of bartonellae in a broad range of bat ectoparasites. Veterinary Microbiology, 159, 541-543.
DOI PMID |
| [11] |
Huson DH, Bryant D (2006) Application of phylogenetic networks in evolutionary studies. Molecular Biology and Evolution, 23, 254-267.
DOI URL |
| [12] |
Janssen BD, Hayes CS (2012) The tmRNA ribosome-rescue system. Advances in Protein Chemistry and Structural Biology, 86, 151-191.
DOI PMID |
| [13] |
Kosoy M, Bai Y, Lynch T, Kuzmin IV, Niezgoda M, Franka R, Agwanda B, Breiman RF, Rupprecht CE (2010) Bartonella spp. in bats, Kenya. Emerging Infectious Diseases, 16, 1875-1881.
DOI URL |
| [14] |
Kosoy M, McKee C, Albayrak L, Fofanov Y (2018) Genotyping of Bartonella bacteria and their animal hosts: Current status and perspectives. Parasitology, 145, 543-562.
DOI PMID |
| [15] |
Kumar S, Stecher G, Li M, Knyaz C, Tamura K (2018) MEGA X: Molecular evolutionary genetics analysis across computing platforms. Molecular Biology and Evolution, 35, 1547-1549.
DOI URL |
| [16] | Kuncova K, Zemlickova H, Jager J, Ryskova L, Pliskova L, Bolehovska R, Pojar M (2019) Diagnosis and treatment of Bartonella endocarditis. Epidemiologie Mikrobiologie Imunologie, 68, 104-108. |
| [17] |
Lei BR, Olival KJ (2014) Contrasting patterns in mammal-bacteria coevolution: Bartonella and Leptospira in bats and rodents. PLoS Neglected Tropical Diseases, 8, e2738.
DOI URL |
| [18] | Li DM, Yu DZ, Liu QY, Gong ZD (2004) Study on the prevalence of Bartonella species in rodent hosts from different enviromental areas in Yunnan. Chinese Journal of Epidemiology, 25, 934-937. (in Chinese with English abstract) |
| [栗冬梅, 俞东征, 刘起勇, 龚正达 (2004) 云南省不同环境鼠形动物巴尔通体感染情况的研究. 中华流行病学杂志, 25, 934-937.] | |
| [19] |
Lin JW, Hsu YM, Chomel BB, Lin LK, Pei JC, Wu SH, Chang CC (2012) Identification of novel Bartonella spp. in bats and evidence of Asian gray shrew as a new potential reservoir of Bartonella. Veterinary Microbiology, 156, 119-126.
DOI URL |
| [20] |
McKee CD, Hayman DTS, Kosoy MY, Webb CT (2016) Phylogenetic and geographic patterns of Bartonella host shifts among bat species. Infection, Genetics and Evolution, 44, 382-394.
DOI URL |
| [21] |
McKee CD, Kosoy MY, Bai Y, Osikowicz LM, Franka R, Gilbert AT, Boonmar S, Rupprecht CE, Peruski LF (2017) Diversity and phylogenetic relationships among Bartonella strains from Thai bats. PLoS ONE, 12, e0181696.
DOI URL |
| [22] |
Milne I, Lindner D, Bayer M, Husmeier D, McGuire G, Marshall DF, Wright F (2009) TOPALi v2: A rich graphical interface for evolutionary analyses of multiple alignments on HPC clusters and multi-core desktops. Bioinformatics, 25, 126-127.
DOI PMID |
| [23] |
Moratelli R, Calisher CH (2015) Bats and zoonotic viruses: Can we confidently link bats with emerging deadly viruses? Memórias Do Instituto Oswaldo Cruz, 110, 1-22.
DOI URL |
| [24] |
Morse SF, Olival KJ, Kosoy M, Billeter S, Patterson BD, Dick CW, Dittmar K (2012) Global distribution and genetic diversity of Bartonella in bat flies (Hippoboscoidea, Streblidae, Nycteribiidae). Infection, Genetics and Evolution, 12, 1717-1723.
DOI URL |
| [25] |
Mühldorfer K (2013) Bats and bacterial pathogens: A review. Zoonoses and Public Health, 60, 93-103.
DOI PMID |
| [26] |
Mühldorfer K, Speck S, Wibbelt G (2011) Diseases in free-ranging bats from Germany. BMC Veterinary Research, 7, 61.
DOI PMID |
| [27] |
Reeves WK, Loftis AD, Gore JA, Dasch GA (2005) Molecular evidence for novel Bartonella species in Trichobius major (Diptera: Streblidae) and Cimex adjunctus (Hemiptera: Cimicidae) from two southeastern bat caves, U.S.A. Journal of Vector Ecology, 30, 339-341.
PMID |
| [28] |
Rodhain F (2015) Bats and viruses: Complex relationships. Bulletin de la Société de Pathologie Exotique, 108, 272-289.
DOI URL |
| [29] |
Rozas J, Ferrer-Mata A, Sánchez-Delbarrio JC, Guirao-Rico S, Librado P, Ramos-Onsins SE, Sánchez-Gracia A (2017) DnaSP 6: DNA sequence polymorphism analysis of large data sets. Molecular Biology and Evolution, 34, 3299-3302.
DOI URL |
| [30] |
Sato S, Brinkerhoff RJ, Hollis E, Funada S, Shannon AB, Maruyama S (2020) Detection of zoonotic Bartonella pathogens in rabbit fleas, Colorado, USA. Emerging Infectious Diseases, 26, 778-781.
DOI URL |
| [31] | Shen B (2014) Molecular evolutionary studies of metabolic genes in bats. PhD dissertation, East China Normal University, Shanghai. (in Chinese with English abstract) |
| [沈斌 (2014) 蝙蝠代谢基因分子进化遗传学研究. 华东师范大学博士学位论文, 上海.] | |
| [32] |
Stuckey MJ, Boulouis HJ, Cliquet F, Picard-Meyer E, Servat A, Aréchiga-Ceballos N, Echevarría JE, Chomel BB (2017) Potentially zoonotic Bartonella in bats from France and Spain. Emerging Infectious Diseases, 23, 539-541.
DOI URL |
| [33] |
Stuckey MJ, Chomel BB de Fleurieu EC, Aguilar-Setién A, Boulouis HJ, Chang CC (2017) Bartonella, bats and bugs: A review. Comparative Immunology, Microbiology and Infectious Diseases, 55, 20-29.
DOI URL |
| [34] | Teeling EC, Springer MS, Madsen O, Bates P, O'Brien SJ, Murphy WJ (2005) A molecular phylogeny for bats illuminates biogeography and the fossil record. Science, 307, 580-584. |
| [35] |
Veikkolainen V, Vesterinen EJ, Lilley TM, Pulliainen AT (2014) Bats as reservoir hosts of human bacterial pathogen, Bartonella mayotimonensis. Emerging Infectious Diseases, 20, 960-967.
DOI PMID |
| [36] | Wang YB, Yang FL, Peng HY, Yang XD, Yang H, Yu BB, Zhang Q, Li ZQ, Yang QJ, Xia ST (2014) Investigation of Bartonella infection in patients with fever of unknown origin in Yunnan Province. China Tropical Medicine, 14, 1446-1448. (in Chinese with English abstract) |
| [王跃兵, 杨发莲, 彭海燕, 杨向东, 杨慧, 于彬彬, 张青, 李志强, 杨秋菊, 夏淑婷 (2014) 云南省不明原因发热病人巴尔通体感染调查. 中国热带医学, 14, 1446-1448.] | |
| [37] |
Wray AK, Olival KJ, Morán D, Lopez MR, Alvarez D, Navarrete-Macias I, Liang E, Simmons NB, Lipkin WI, Daszak P, Anthony SJ (2016) Viral diversity, prey preference, and Bartonella prevalence in Desmodus rotundus in Guatemala. EcoHealth, 13, 761-774.
PMID |
| [1] | 贾晓旭, 陈皖强, 唐修君, 樊艳凤, 张静, 王海威, 高玉时. 西南地区家鸡线粒体DNA控制区遗传多样性和基因渗入[J]. 生物多样性, 2026, 34(5): 26003-. |
| [2] | 姜媛, 黄贝希, 贾学圆, 梁思, 谢雨彤, 范平, 宋刚. 一种基于不等长DNA序列的单倍型丰富度估算方法[J]. 生物多样性, 2026, 34(1): 25263-. |
| [3] | 王儒晓, 史博洋, 潘达, 孙红英. 中国特有华溪蟹属淡水蟹多样性格局及其保护空缺[J]. 生物多样性, 2025, 33(8): 25123-. |
| [4] | 周智成, 曹天玲, 刘如垚, 丁琪琪, 马轲, 杨丽萍, 周传江, 聂国兴, 汤永涛. 基于线粒体COI基因的黄河流域麦穗鱼种群遗传多样性与遗传结构[J]. 生物多样性, 2025, 33(8): 24501-. |
| [5] | 王蔼英, 廖万金. 协同演化研究: 协同系统发育分析方法与进展[J]. 生物多样性, 2025, 33(8): 25112-. |
| [6] | 范平, 王欢, 温知新, 宋刚, 雷富民. 气候因子对鸟类遗传多样性与物种分布面积关系的影响[J]. 生物多样性, 2025, 33(8): 25072-. |
| [7] | 薛瑞翔, 马雪蓉, 吴炯文, 刘爱君, 张细权, 季从亮, 殷颖珊, 朱炜健, 罗庆斌. 中山麻鸭群体遗传多样性与遗传结构[J]. 生物多样性, 2025, 33(8): 24592-. |
| [8] | 范平, 温知新, 宋刚. 气候因子和人类活动对两栖及哺乳动物不同遗传多样性指标的影响[J]. 生物多样性, 2025, 33(8): 25022-. |
| [9] | 廖雅晴, 黄泽锋, 王晓云, 张礼标, 吴毅, 余文华. 广东省翼手目物种名录更新及分子条形码数据库[J]. 生物多样性, 2025, 33(4): 24584-. |
| [10] | 伍金山, 杨长乐, 马玉凤, 李亚旋, 高文家, 叶·库斯力, 叶樑洪, 杨宇骄, 徐梦琦, 廖廷琼, 钟林强, 单文娟. 艾比湖湿地国家级自然保护区马鹿遗传多样性及遗传结构[J]. 生物多样性, 2025, 33(12): 25233-. |
| [11] | 王嘉陈, 徐汤俊, 许唯, 张高季, 尤艺瑾, 阮宏华, 刘宏毅. 城市景观格局对大蚰蜒种群遗传结构的影响[J]. 生物多样性, 2025, 33(1): 24251-. |
| [12] | 耿江天, 王菲, 赵华斌. 城市化对中国蝙蝠影响的研究进展[J]. 生物多样性, 2024, 32(8): 24109-. |
| [13] | 刘莹莹, 龚立新, 曾皓, 冯江, 董永军, 王磊, 江廷磊. 被动声学监测在蝙蝠研究中的应用[J]. 生物多样性, 2024, 32(10): 24233-. |
| [14] | 冯晨, 张洁, 黄宏文. 统筹植物就地保护与迁地保护的解决方案: 植物并地保护(parallel situ conservation)[J]. 生物多样性, 2023, 31(9): 23184-. |
| [15] | 李庆多, 栗冬梅. 全球蝙蝠巴尔通体流行状况分析[J]. 生物多样性, 2023, 31(9): 23166-. |
| 阅读次数 | ||||||
|
全文 |
|
|||||
|
摘要 |
|
|||||
备案号:京ICP备16067583号-7
Copyright © 2026 版权所有 《生物多样性》编辑部
地址: 北京香山南辛村20号, 邮编:100093
电话: 010-62836137, 62836665 E-mail: biodiversity@ibcas.ac.cn
![]()