
生物多样性 ›› 2019, Vol. 27 ›› Issue (5): 526-533. DOI: 10.17520/biods.2018209 cstr: 32101.14.biods.2018209
所属专题: 物种形成与系统进化
刘山林( )
)
收稿日期:2018-07-30
接受日期:2018-12-25
出版日期:2019-05-20
发布日期:2019-05-20
通讯作者:
刘山林
基金资助:
Liu Shanlin()
Received:2018-07-30
Accepted:2018-12-25
Online:2019-05-20
Published:2019-05-20
Contact:
Liu Shanlin
摘要:
近年来DNA条形码技术迅速发展, 产生的条形码的数量及其应用范围都呈指数性增长, 现已广泛用于物种鉴定、食性分析、生物多样性评估等方面。本文重点总结并讨论了构建条形码参考数据库和序列聚类相关的信息分析的技术和方法, 包括: 基于高通量测序(high throughput sequencing, HTS)平台以高效并较低的成本获取条形码序列的方法; 同时还介绍了从原始测序序列到分类操作单元(operational taxonomic units, OTUs)过程中的一些计算逻辑以及被广泛采用的软件和技术。这是一个较新并快速发展的领域, 我们希望本文能为读者提供一个梗概, 了解DNA条形码技术在生物多样性研究应用中的方法和手段。
刘山林 (2019) DNA条形码参考数据集构建和序列分析相关的新兴技术. 生物多样性, 27, 526-533. DOI: 10.17520/biods.2018209.
Liu Shanlin (2019) DNA barcoding and emerging reference construction and data analysis technologies. Biodiversity Science, 27, 526-533. DOI: 10.17520/biods.2018209.
| 标记基因 Marker gene | 目标物种 Targeted group | 数据库 Database |
|---|---|---|
| 16S | 细菌和古细菌 Bacteria and archea ( | 核糖体数据库项目 Ribosomal Database Project ( |
| ITS | 真菌( Fungi ( | UNITE ( |
| 18S | 原生生物 Protist ( | SILVA ( |
| matK + rbcL | 植物 Plant ( | 生命条形码数据库 Barcode of Life Data Systems ( |
| COI | 动物群( Fauna ( | 核糖体数据库项目 Ribosomal Database Project ( |
表1 广泛用于DNA条形码技术的标记基因
Table 1 Marker genes widely used for barcoding
| 标记基因 Marker gene | 目标物种 Targeted group | 数据库 Database |
|---|---|---|
| 16S | 细菌和古细菌 Bacteria and archea ( | 核糖体数据库项目 Ribosomal Database Project ( |
| ITS | 真菌( Fungi ( | UNITE ( |
| 18S | 原生生物 Protist ( | SILVA ( |
| matK + rbcL | 植物 Plant ( | 生命条形码数据库 Barcode of Life Data Systems ( |
| COI | 动物群( Fauna ( | 核糖体数据库项目 Ribosomal Database Project ( |
| 目标序列长度 Targeted region length (bp) | 优势 Advantages | 劣势 Disadvantages | 参考文献 Reference |
|---|---|---|---|
| ~300 | - | 无法处理较长的目标序列; Roche 454平台 Can not work on long fragments; Roche 454 platform | Shokralla et al, 2014 |
| ~180 | 简单, 易操作, 成本低 Straightforward, easy to operate, cost-efficient | 目标序列偏短, 只能用于物种初筛 Short targeted region; can only be used for species pre-clustering | Meier et al, 2016 |
| ~650 | 标准DNA条形码全长 Standard full-length COI | 普适性差; 需要多轮PCR过程 Poor universality; multiple rounds of PCR | Shokralla et al, 2015; Cruaud et al, 2017 |
| ~650 | 易操作, 标准DNA条形码全长 Easy to operate, standard full-length COI | 相对较高的计算资源 Relatively high requirement for computational resources | Liu et al, 2017 |
| ~650 | 易操作, 标准DNA条形码全长 Easy to operate, standard full-length COI | SMRT平台成本高 High cost of SMRT platform | Hebert et al, 2018 |
| ~650 | 易操作, 标准DNA条形码全长 Easy to operate, standard full-length COI | 测序平台暂时不够普及 Not a mass production | Yang et al, 2018 |
表2 利用高通量测序平台批量获取DNA条形码的方法
Table 2 High throughput methods to achieve barcode sequences
| 目标序列长度 Targeted region length (bp) | 优势 Advantages | 劣势 Disadvantages | 参考文献 Reference |
|---|---|---|---|
| ~300 | - | 无法处理较长的目标序列; Roche 454平台 Can not work on long fragments; Roche 454 platform | Shokralla et al, 2014 |
| ~180 | 简单, 易操作, 成本低 Straightforward, easy to operate, cost-efficient | 目标序列偏短, 只能用于物种初筛 Short targeted region; can only be used for species pre-clustering | Meier et al, 2016 |
| ~650 | 标准DNA条形码全长 Standard full-length COI | 普适性差; 需要多轮PCR过程 Poor universality; multiple rounds of PCR | Shokralla et al, 2015; Cruaud et al, 2017 |
| ~650 | 易操作, 标准DNA条形码全长 Easy to operate, standard full-length COI | 相对较高的计算资源 Relatively high requirement for computational resources | Liu et al, 2017 |
| ~650 | 易操作, 标准DNA条形码全长 Easy to operate, standard full-length COI | SMRT平台成本高 High cost of SMRT platform | Hebert et al, 2018 |
| ~650 | 易操作, 标准DNA条形码全长 Easy to operate, standard full-length COI | 测序平台暂时不够普及 Not a mass production | Yang et al, 2018 |
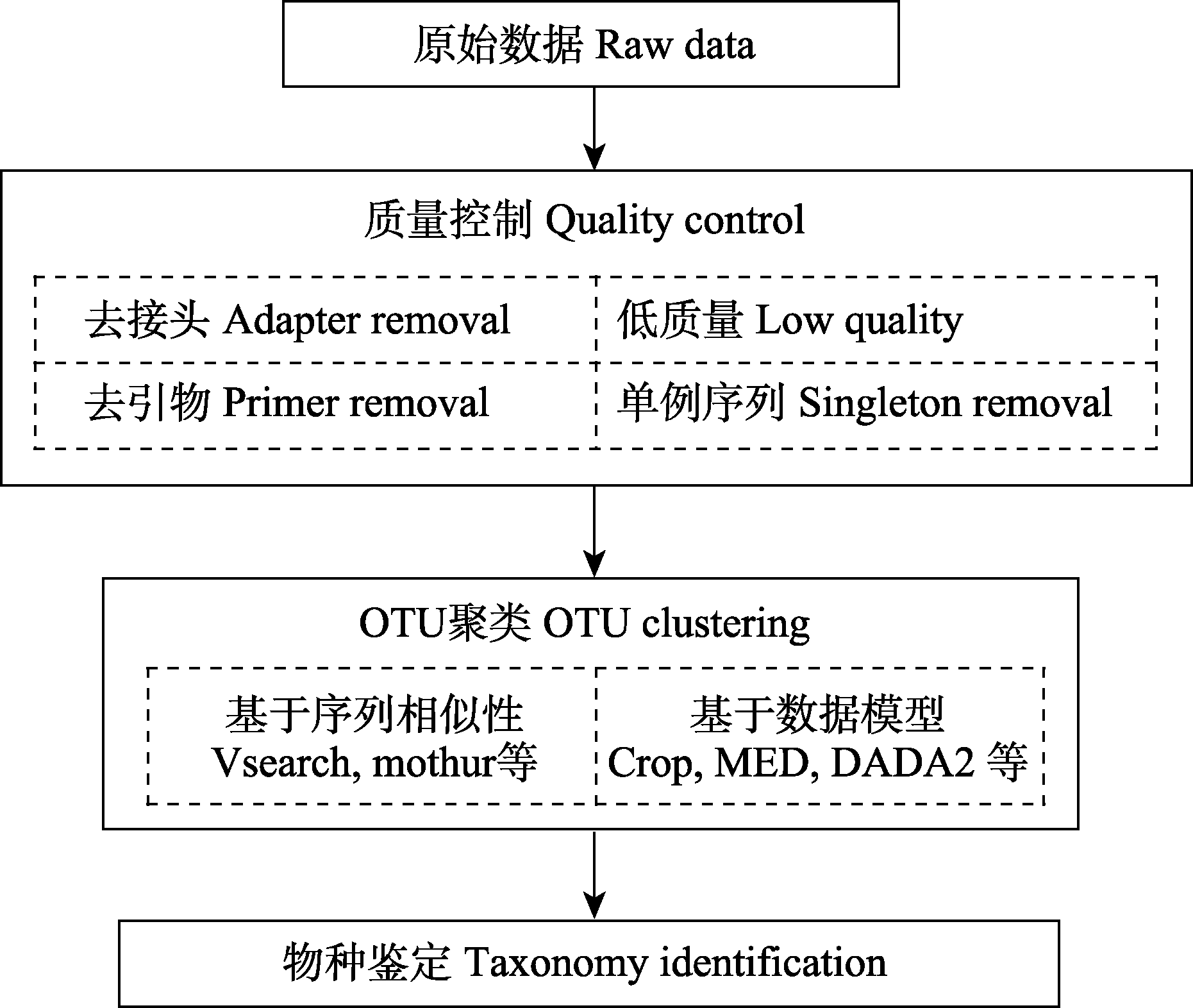
图1 条形码分析的数据处理流程图
Fig. 1 Diagram of DNA barcode data analysis
| [1] |
Armstrong K, Ball S ( 2005) DNA barcodes for biosecurity: Invasive species identification. Philosophical Transactions of the Royal Society of London B: Biological Sciences, 360, 1813-1823.
DOI URL |
| [2] |
Baird DJ, Hajibabaei M ( 2012) Biomonitoring 2.0: A new paradigm in ecosystem assessment made possible by next-generation DNA sequencing. Molecular Ecology, 21, 2039-2044.
DOI URL |
| [3] |
Benson DA, Cavanaugh M, Clark K, Karsch-Mizrachi I, Lipman DJ, Ostell J, Sayers EW ( 2012) GenBank. Nucleic Acids Research, 41, D36-D42.
DOI URL |
| [4] | Bohmann K, Evans A, Gilbert MTP, Carvalho GR, Creer S, Knapp M, Douglas WY, De Bruyn M ( 2014) Environmental DNA for wildlife biology and biodiversity monitoring. Trends in Ecology & Evolution, 29, 358-367. |
| [5] |
Bohmann K, Schnell IB, Gilbert MTP ( 2013) When bugs reveal biodiversity. Molecular Ecology, 22, 909-911.
DOI URL |
| [6] |
Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP ( 2016) DADA2: High-resolution sample inference from Illumina amplicon data. Nature Methods, 13, 581-583.
DOI |
| [7] |
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI ( 2010) QIIME allows analysis of high-throughput community sequencing data. Nature Methods, 7, 335-336.
DOI |
| [8] |
Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Lozupone CA, Turnbaugh PJ, Fierer N, Knight R ( 2011) Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proceedings of the National Academy of Sciences, USA, 108, 4516-4522.
DOI URL |
| [9] | Chen Y, Chen Y, Shi C, Huang Z, Zhang Y, Li S, Li Y, Ye J, Yu C, Li Z ( 2017) SOAPnuke: A MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. GigaScience, 7, gix120. |
| [10] | Cole JR, Wang Q, Cardenas E, Fish J, Chai B, Farris RJ, Kulam-Syed-Mohideen A, McGarrell DM, Marsh T, Garrity GM ( 2008) The Ribosomal Database Project: Improved alignments and new tools for rRNA analysis. Nucleic Acids Research, 37, D141-D145. |
| [11] |
Cruaud P, Rasplus J-Y, Rodriguez LJ, Cruaud A ( 2017) High-throughput sequencing of multiple amplicons for barcoding and integrative taxonomy. Scientific Reports, 7, 41948.
DOI |
| [12] |
Deiner K, Bik HM, Mächler E, Seymour M, Lacoursière-Roussel A, Altermatt F, Creer S, Bista I, Lodge DM, Vere N ( 2017) Environmental DNA metabarcoding: Transforming how we survey animal and plant communities. Molecular Ecology, 26, 5872-5895.
DOI URL |
| [13] | DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, Dalevi D, Hu P, Andersen GL ( 2006) Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Applied and Environmental Microbiology 72, 5069-5072. |
| [14] |
Edgar RC ( 2010) Search and clustering orders of magnitude faster than BLAST. Bioinformatics, 26, 2460-2461.
DOI URL |
| [15] |
Edgar RC ( 2013) UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nature Methods, 10, 996-998.
DOI |
| [16] |
Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R ( 2011) UCHIME improves sensitivity and speed of chimera detection. Bioinformatics, 27, 2194-2200.
DOI URL |
| [17] | Eid J, Fehr A, Gray J, Luong K, Lyle J, Otto G, Peluso P, Rank D, Baybayan P, Bettman B ( 2008) Real-time DNA sequencing from single polymerase molecules. Science, 323, 133-138. |
| [18] |
Eren AM, Morrison HG, Lescault PJ, Reveillaud J, Vineis JH, Sogin ML ( 2015) Minimum entropy decomposition: Unsupervised oligotyping for sensitive partitioning of high-throughput marker gene sequences. The ISME Journal, 9, 968-979.
DOI |
| [19] |
Frøslev TG, Kjøller R, Bruun HH, Ejrnæs R, Brunbjerg AK, Pietroni C, Hansen AJ ( 2017) Algorithm for post-clustering curation of DNA amplicon data yields reliable biodiversity estimates. Nature Communications, 8, 1188.
DOI URL |
| [20] |
Group CPB, Li DZ, Gao LM, Li HT, Wang H, Ge XJ, Liu JQ, Chen ZD, Zhou SL, Chen SL ( 2011) Comparative analysis of a large dataset indicates that internal transcribed spacer (ITS) should be incorporated into the core barcode for seed plants. Proceedings of the National Academy of Sciences, USA, 108, 19641-19646.
DOI URL |
| [21] |
Hajibabaei M, Baird DJ, Fahner NA, Beiko R, Golding GB ( 2016) A new way to contemplate Darwin’s tangled bank: How DNA barcodes are reconnecting biodiversity science and biomonitoring. Philosophical Transactions of the Royal Society of London B: Biological Sciences, 371, 20150330.
DOI URL |
| [22] |
Hao X, Jiang R, Chen T ( 2011) Clustering 16S rRNA for OTU prediction: A method of unsupervised Bayesian clustering. Bioinformatics, 27, 611-618.
DOI URL |
| [23] |
Hebert PD, Braukmann TW, Prosser SW, Ratnasingham S, Ivanova NV, Janzen DH, Hallwachs W, Naik S, Sones JE, Zakharov EV ( 2018) A Sequel to Sanger: Amplicon sequencing that scales. BMC Genomics, 19, 219.
DOI |
| [24] |
Hebert PD, Cywinska A, Ball SL ( 2003) Biological identifications through DNA barcodes. Proceedings of the Royal Society of London B: Biological Sciences, 270, 313-321.
DOI URL |
| [25] |
Hebert PD, Hollingsworth PM, Hajibabaei M ( 2016) From writing to reading the encyclopedia of life. Proceedings of the Royal Society of London B: Biological Sciences, 371, 20150321.
DOI URL |
| [26] |
Hollingsworth PM, Forrest LL, Spouge JL, Hajibabaei M, Ratnasingham S, van der Bank M, Chase MW, Cowan RS, Erickson DL ( 2009) A DNA barcode for land plants. Proceedings of the National Academy of Sciences, USA, 106, 12794-12797.
DOI URL |
| [27] |
Jiang H, Lei R, Ding SW, Zhu S ( 2014) Skewer: A fast and accurate adapter trimmer for next-generation sequencing paired-end reads. BMC Bioinformatics, 15, 182.
DOI |
| [28] |
Kõljalg U, Larsson KH, Abarenkov K, Nilsson RH, Alexander IJ, Eberhardt U, Erland S, Høiland K, Kjøller R, Larsson E ( 2005) UNITE: A database providing web-based methods for the molecular identification of ectomycorrhizal fungi. New Phytologist, 166, 1063-1068.
DOI URL |
| [29] | Kress WJ, Erickson DL ( 2012) DNA barcodes: Methods and protocols. In: DNA Barcodes (eds Kress WJ, Erickson DL), pp. 3-8.Humana Press, Totowa. |
| [30] |
Kunz TH, Whitaker JO Jr ( 1983) An evaluation of fecal analysis for determining food habits of insectivorous bats. Canadian Journal of Zoology, 61, 1317-1321.
DOI URL |
| [31] |
Liu S, Li Y, Lu J, Su X, Tang M, Zhang R, Zhou L, Zhou C, Yang Q, Ji Y ( 2013) SOAPBarcode: Revealing arthropod biodiversity through assembly of Illumina shotgun sequences of PCR amplicons. Methods in Ecology and Evolution, 4, 1142-1150.
DOI URL |
| [32] |
Liu S, Wang X, Xie L, Tan M, Li Z, Su X, Zhang H, Misof B, Kjer KM, Tang M ( 2016) Mitochondrial capture enriches mito-DNA 100 fold, enabling PCR-free mitogenomics biodiversity analysis. Molecular Ecology Resources, 16, 470-479.
DOI URL |
| [33] | Liu S, Yang C, Zhou C, Zhou X ( 2017) Filling reference gaps via assembling DNA barcodes using high-throughput sequencing—Moving toward barcoding the world. GigaScience, 6, 1-8. |
| [34] |
Mahon AR, Jerde CL, Galaska M, Bergner JL, Chadderton WL, Lodge DM, Hunter ME, Nico LG ( 2013) Validation of eDNA surveillance sensitivity for detection of Asian carps in controlled and field experiments. PLoS ONE, 8, e58316.
DOI URL |
| [35] | Matias Rodrigues JF, von Mering C ( 2013) HPC-CLUST: Distributed hierarchical clustering for large sets of nucleotide sequences. Bioinformatics, 30, 287-288. |
| [36] |
Meier R, Wong W, Srivathsan A, Foo M ( 2016) $1 DNA barcodes for reconstructing complex phenomes and finding rare species in specimen-rich samples. Cladistics, 32, 100-110.
DOI URL |
| [37] |
Nilsson RH, Ryberg M, Abarenkov K, Sjökvist E, Kristiansson E ( 2009) The ITS region as a target for characterization of fungal communities using emerging sequencing technologies. FEMS Microbiology Letters, 296, 97-101.
DOI URL |
| [38] |
Pawlowski J, Audic S, Adl S, Bass D, Belbahri L, Berney C, Bowser SS, Cepicka I, Decelle J, Dunthorn M ( 2012) CBOL protist working group: Barcoding eukaryotic richness beyond the animal, plant, and fungal kingdoms. PLoS Biology, 10, e1001419.
DOI URL |
| [39] |
Pruesse E, Quast C, Knittel K, Fuchs BM, Ludwig W, Peplies J, Glöckner FO ( 2007) SILVA: A comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Research, 35, 7188-7196.
DOI URL |
| [40] |
Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glöckner FO ( 2012) The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Research, 41, D590-D596.
DOI URL |
| [41] | Ratnasingham S, Hebert PD (2007)BOLD: The Barcode of Life Data System (.Molecular Ecology Notes, 7, 355-364. |
| [42] |
Rognes T, Flouri T, Nichols B, Quince C, Mahé F ( 2016) VSEARCH: A versatile open source tool for metagenomics. PeerJ, 4, e2584.
DOI URL |
| [43] |
Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ ( 2009) Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Applied and Environmental Microbiology, 75, 7537-7541.
DOI URL |
| [44] |
Schnell IB, Thomsen PF, Wilkinson N, Rasmussen M, Jensen LRD, Willerslev E, Bertelsen MF, Gilbert MTP ( 2012) Screening mammal biodiversity using DNA from leeches. Current Biology, 22, R262-R263.
DOI URL |
| [45] |
Schoch CL, Seifert KA, Huhndorf S, Robert V, Spouge JL, Levesque CA, Chen W, Bolchacova E, Voigt K, Crous PW ( 2012) Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for fungi. Proceedings of the National Academy of Sciences, USA, 109, 6241-6246.
DOI URL |
| [46] |
Schubert M, Lindgreen S, Orlando L ( 2016) AdapterRemoval v2: Rapid adapter trimming, identification, and read merging. BMC Research Notes, 9, 88.
DOI URL |
| [47] |
Shi ZY, Yang CQ, Hao MD, Wang XY, Ward RD, Zhang AB ( 2018) FuzzyID2: A software package for large data set species identification via barcoding and metabarcoding using hidden Markov models and fuzzy set methods. Molecular Ecology Resources, 18, 666-675.
DOI URL |
| [48] | Shokralla S, Gibson JF, Nikbakht H, Janzen DH, Hallwachs W, Hajibabaei M ( 2014) Next-generation DNA barcoding: Using next-generation sequencing to enhance and accelerate DNA barcode capture from single specimens. Molecular Ecology Resources, 14, 892-901. |
| [49] |
Shokralla S, Porter TM, Gibson JF, Dobosz R, Janzen DH, Hallwachs W, Golding GB, Hajibabaei M ( 2015) Massively parallel multiplex DNA sequencing for specimen identification using an Illumina MiSeq platform. Scientific Reports, 5, 9687.
DOI |
| [50] |
Sogin ML, Morrison HG, Huber JA, Welch DM, Huse SM, Neal PR, Arrieta JM, Herndl GJ ( 2006) Microbial diversity in the deep sea and the underexplored “rare biosphere”. Proceedings of the National Academy of Sciences, USA, 103, 12115-12120.
DOI URL |
| [51] |
Taberlet P, Coissac E, Hajibabaei M, Rieseberg LH ( 2012) Environmental DNA. Molecular Ecology, 21, 1789-1793.
DOI URL |
| [52] |
Tang M, Hardman CJ, Ji Y, Meng G, Liu S, Tan M, Yang S, Moss ED, Wang J, Yang C ( 2015) High-throughput monitoring of wild bee diversity and abundance via mitogenomics. Methods in Ecology and Evolution, 6, 1034-1043.
DOI URL |
| [53] |
Turner CR, Miller DJ, Coyne KJ, Corush J ( 2014) Improved methods for capture, extraction, and quantitative assay of environmental DNA from Asian bigheaded carp (Hypophthalmichthys spp.). PLoS ONE, 9, e114329.
DOI URL |
| [54] | Yang C, Tan S, Meng G, Bourne DG, O’Brien PA, Xu J, Liao S, Chen A, Chen X, Liu S ( 2018) Access COI barcode efficiently using high throughput Single End 400 bp sequencing. bioRxiv, doi: 10.1101/498618 . |
| [55] |
Yu DW, Ji YQ, Emerson BC, Wang XY, Ye CX, Yang CY, Ding ZL ( 2012) Biodiversity soup: Metabarcoding of arthropods for rapid biodiversity assessment and biomonitoring. Methods in Ecology and Evolution, 3, 613-623.
DOI URL |
| [56] |
Zhang J, Kapli P, Pavlidis P, Stamatakis A ( 2013) A general species delimitation method with applications to phylogenetic placements. Bioinformatics, 29, 2869-2876.
DOI URL |
| [57] |
Zhou X, Li Y, Liu S, Yang Q, Su X, Zhou L, Tang M, Fu R, Li J, Huang Q ( 2013) Ultra-deep sequencing enables high-fidelity recovery of biodiversity for bulk arthropod samples without PCR amplification. GigaScience, 2, 4.
DOI URL |
| [1] | 尚华丹, 张楚晴, 王梅, 裴文娅, 李国宏, 王鸿斌. 中国杨树害虫物种多样性及其地理分布[J]. 生物多样性, 2025, 33(2): 24370-. |
| [2] | 吴相獐, 雷富民, 单壹壹, 于晶. 上海城市公园苔藓植物多样性分布格局及其环境影响因子[J]. 生物多样性, 2024, 32(2): 23364-. |
| [3] | 罗正明, 刘晋仙, 张变华, 周妍英, 郝爱华, 杨凯, 柴宝峰. 不同退化阶段亚高山草甸土壤原生生物群落多样性特征及驱动因素[J]. 生物多样性, 2023, 31(8): 23136-. |
| [4] | 罗小燕, 李强, 黄晓磊. 戴云山国家级自然保护区访花昆虫DNA条形码数据集[J]. 生物多样性, 2023, 31(8): 23236-. |
| [5] | 邢超, 林依, 周智强, 赵联军, 蒋仕伟, 林蓁蓁, 徐基良, 詹祥江. 基于DNA条形码技术构建王朗国家级自然保护区陆生脊椎动物遗传资源数据库及物种鉴定[J]. 生物多样性, 2023, 31(7): 22661-. |
| [6] | 毛莹儿, 周秀梅, 王楠, 李秀秀, 尤育克, 白尚斌. 毛竹扩张对杉木林土壤细菌群落的影响[J]. 生物多样性, 2023, 31(6): 22659-. |
| [7] | 吴帆, 刘深云, 江虎强, 王茜, 陈开威, 李红亮. 中华蜜蜂和意大利蜜蜂秋冬期传粉植物多样性比较[J]. 生物多样性, 2023, 31(5): 22528-. |
| [8] | 赵雯, 王丹丹, 热依拉·木民, 黄开钏, 刘顺, 崔宝凯. 阿尔山地区兴安落叶松林土壤微生物群落结构[J]. 生物多样性, 2023, 31(2): 22258-. |
| [9] | 吴科毅, 阮文达, 周棣锋, 陈庆春, 张承云, 潘新园, 余上, 刘阳, 肖荣波. 基于音节聚类分析的被动声学监测技术及其在鸟类监测中的应用[J]. 生物多样性, 2023, 31(1): 22370-. |
| [10] | 夏凡, 杨婧, 李建, 史洋, 盖立新, 黄文华, 张经纬, 杨南, 高福利, 韩莹莹, 鲍伟东. 北京地区四个豹猫亚种群肠道菌群的组成[J]. 生物多样性, 2022, 30(9): 22103-. |
| [11] | 孙翌昕, 李英滨, 李玉辉, 李冰, 杜晓芳, 李琪. 高通量测序技术在线虫多样性研究中的应用[J]. 生物多样性, 2022, 30(12): 22266-. |
| [12] | 高程, 郭良栋. 微生物物种多样性、群落构建与功能性状研究进展[J]. 生物多样性, 2022, 30(10): 22429-. |
| [13] | 夏呈强, 李毅, 党延茹, 察倩倩, 贺晓艳, 秦启龙. 中印度洋与南海西部表层海水细菌多样性[J]. 生物多样性, 2022, 30(1): 21407-. |
| [14] | 俞正森, 宋娜, 本村浩之, 高天翔. 中国银口天竺鲷属鱼类的分类厘定[J]. 生物多样性, 2021, 29(7): 971-979. |
| [15] | 陆奇丰, 黄至欢, 骆文华. 极小种群濒危植物广西火桐、丹霞梧桐的叶绿体基因组特征[J]. 生物多样性, 2021, 29(5): 586-595. |
| 阅读次数 | ||||||
|
全文 |
|
|||||
|
摘要 |
|
|||||
备案号:京ICP备16067583号-7
Copyright © 2026 版权所有 《生物多样性》编辑部
地址: 北京香山南辛村20号, 邮编:100093
电话: 010-62836137, 62836665 E-mail: biodiversity@ibcas.ac.cn
![]()