
生物多样性 ›› 2026, Vol. 34 ›› Issue (1): 25263. DOI: 10.17520/biods.2025263 cstr: 32101.14.biods.2025263
姜媛1, 黄贝希1, 贾学圆1, 梁思1, 谢雨彤1, 范平1,*( )(
)( ), 宋刚2()
), 宋刚2()
收稿日期:2025-07-06
接受日期:2025-09-30
出版日期:2026-01-20
发布日期:2026-01-22
通讯作者:
范平
基金资助:
Yuan Jiang1, Beixi Huang1, Xueyuan Jia1, Si Liang1, Yutong Xie1, Ping Fan1,*()(), Gang Song2()
Received:2025-07-06
Accepted:2025-09-30
Online:2026-01-20
Published:2026-01-22
Contact:
Ping Fan
Supported by:摘要:
传统遗传多样性计算方法要求序列长度一致, 而从公共数据库中获取的序列长度不一, 增加了单倍型识别和遗传多样性评估的难度。现有方法虽能估算不等长DNA序列的单倍型多样性和核苷酸多样性, 但单倍型丰富度的有效计算方法仍属空白。为此, 本研究基于配对序列间的核苷酸差异(Kij)开发了一种针对不等长DNA序列的单倍型丰富度估算方法。我们通过3项分析验证方法性能: (1)对于同一套等长序列数据集, 将其计算结果与DnaSP的输出结果进行比较; (2)基于鸟类、哺乳动物和两栖动物的等长数据集生成随机长度的模拟序列, 验证算法处理不等长序列的性能, 并利用药用植物数据集评估该方法的泛化能力; (3)应用该方法计算了鸟类、哺乳动物和两栖动物单倍型丰富度的纬度梯度格局。结果表明: (1)对等长序列, 新方法与DnaSP的输出结果无显著差异(鸟类: W = 22,018, P = 0.845; 哺乳动物: W = 23,096, P = 0.990; 两栖动物: W = 3,518.5, P = 0.977), 且在存在碱基缺失时展现出更优的单倍型识别能力(平均较DnaSP多识别1.333 ± 0.188个单倍型); (2)随机长度模拟实验证实该方法对不等长的序列数据具备良好的估算性能(以相对误差衡量的整体计算精确度为0.130 ± 0.106; 稳定性为0.007 ± 0.007); (3)纬度格局分析显示: 鸟类和哺乳动物在南半球从北到南呈现显著递减趋势, 而在北半球的递减趋势较为平缓; 两栖动物则呈现自南向北的持续递减模式。本研究有助于开发更精确的量化方法, 或可为遗传多样性研究及保护工作提供新的分析工具。
姜媛, 黄贝希, 贾学圆, 梁思, 谢雨彤, 范平, 宋刚 (2026) 一种基于不等长DNA序列的单倍型丰富度估算方法. 生物多样性, 34, 25263. DOI: 10.17520/biods.2025263.
Yuan Jiang, Beixi Huang, Xueyuan Jia, Si Liang, Yutong Xie, Ping Fan, Gang Song (2026) An approach for estimating haplotype richness from DNA sequences with unequal lengths. Biodiversity Science, 34, 25263. DOI: 10.17520/biods.2025263.
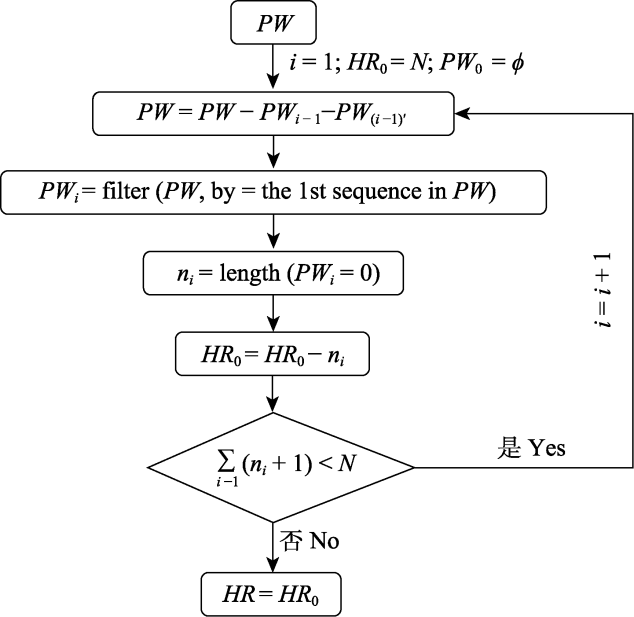
图1 单倍型丰富度计算流程
Fig. 1 The computational framework of haplotype richness
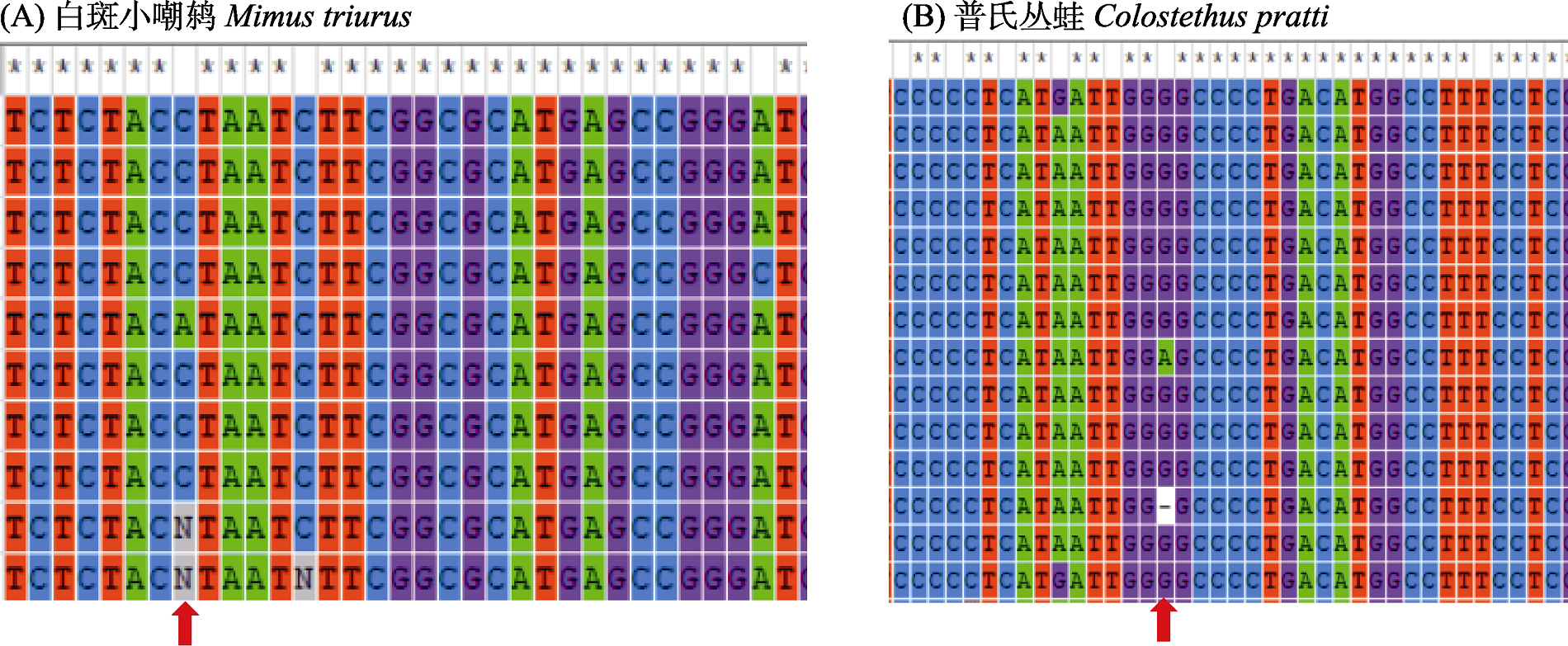
图2 存在碱基缺失的等长序列物种数据示例(局部数据)。红色箭头标注了存在缺失碱基信号的核苷酸分离位点。
Fig. 2 An example of species data for sequences of equal length with missing bases (partial range). Red arrows indicate the nucleotide segregation sites where missing bases are found.
| 伪R2 Pseudo R2 | 一次项系数 β1 | 二次项系数 β2 | |
|---|---|---|---|
| 鸟类 Birds | 0.280 | 1.458e-02*** | -5.116e-04*** |
| 哺乳动物 Mammals | 0.361 | 1.788e-02*** | -5.506e-04*** |
| 两栖动物 Amphibians | 0.518 | -8.046e-02*** | 4.659e-04*** |
表1 鸟类、哺乳动物和两栖动物单倍型丰富度纬度梯度的beta回归模型
Table 1 Beta regression models for latitudinal gradients in haplotype richness of birds, mammals, and amphibians
| 伪R2 Pseudo R2 | 一次项系数 β1 | 二次项系数 β2 | |
|---|---|---|---|
| 鸟类 Birds | 0.280 | 1.458e-02*** | -5.116e-04*** |
| 哺乳动物 Mammals | 0.361 | 1.788e-02*** | -5.506e-04*** |
| 两栖动物 Amphibians | 0.518 | -8.046e-02*** | 4.659e-04*** |
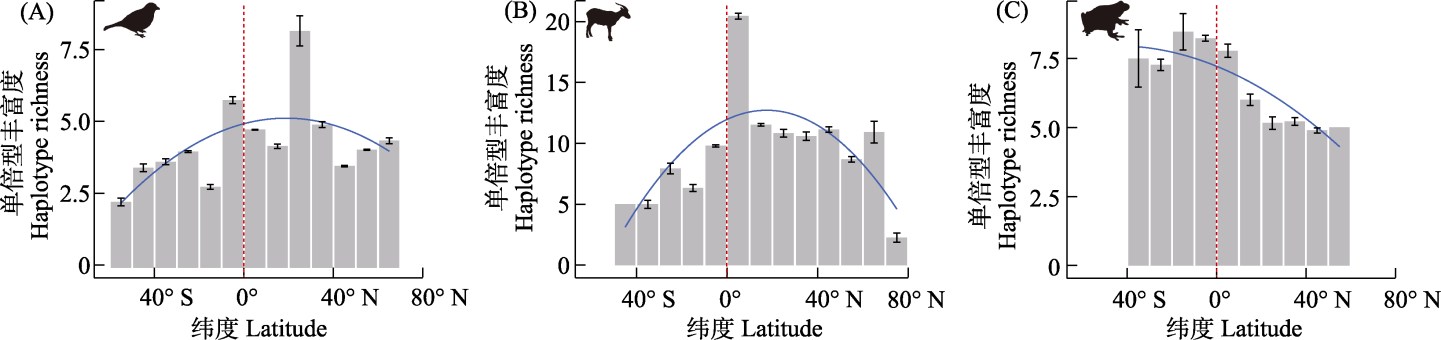
图3 鸟类(A)、哺乳动物(B)和两栖动物(C)单倍型丰富度的纬度梯度格局。蓝色回归线代表beta回归模型的预测结果。红色虚线标示赤道位置。
Fig. 3 The latitudinal gradients in haplotype richness of birds (A), mammals (B), and amphibians (C). The regression lines (blue lines) represent the predictions of the beta regression model. The red dashed lines show the equator.
| [1] |
Chang YB, Song G, Zhang DZ, Jia CX, Fan P, Hao Y, Ji YZ, Lei FM (2022) Distribution pattern and driving factors of genetic diversity of passerine birds in the mountains of Southwest China. Avian Research, 13, 100043.
DOI URL |
| [2] |
Edgar RC (2022) Muscle5: High-accuracy alignment ensembles enable unbiased assessments of sequence homology and phylogeny. Nature Communications, 13, 6968.
DOI PMID |
| [3] |
Ellegren H, Galtier N (2016) Determinants of genetic diversity. Nature Reviews Genetics, 17, 422-433.
DOI PMID |
| [4] |
Fan P, Fjeldså J, Liu X, Dong YF, Chang YB, Qu YH, Song G, Lei FM (2021) An approach for estimating haplotype diversity from sequences with unequal lengths. Methods in Ecology and Evolution, 12, 1658-1667.
DOI URL |
| [5] |
Fan P, Song G, Qiao HJ, Zhang DZ, Ji YZ, Qu YH, Fjeldså J, Lei FM (2025) Revaluation of the genetic diversity-area relationship by integrating nucleotide and haplotype diversity. Current Zoology, 71, 645-651.
DOI URL |
| [6] |
Ferrari S, Cribari-Neto F (2004) Beta regression for modelling rates and proportions. Journal of Applied Statistics, 31, 799-815.
DOI URL |
| [7] |
Fordham DA, Saltré F, Haythorne S, Wigley TML, Otto- Bliesner BL, Chan KC, Brook BW (2017) PaleoView: A tool for generating continuous climate projections spanning the last 21000 years at regional and global scales. Ecography, 40, 1348-1358.
DOI URL |
| [8] |
Goodall-Copestake WP, Tarling GA, Murphy EJ (2012) On the comparison of population-level estimates of haplotype and nucleotide diversity: A case study using the gene cox1 in animals. Heredity, 109, 50-56.
DOI PMID |
| [9] |
Gratton P, Marta S, Bocksberger G, Winter M, Keil P, Trucchi E, Kühl H (2017) Which latitudinal gradients for genetic diversity? Trends in Ecology & Evolution, 32, 724-726.
DOI URL |
| [10] | Hijmans RJ, Williams E, Vennes C (2019) geosphere: Spherical Trigonometry. R Package Version 1.5-10, https://CRAN.R-project.org/package=geosphere. (accessed on 2021-05-21) |
| [11] |
Hong B, Rabassa J, Uchida M, Hong YT, Peng HJ, Ding HW, Guo Q, Yao H (2019) Response and feedback of the Indian summer monsoon and the Southern Westerly Winds to a temperature contrast between the hemispheres during the last glacial-interglacial transitional period. Earth-Science Reviews, 197, 102917.
DOI URL |
| [12] |
Leitwein M, Duranton M, Rougemont Q, Gagnaire PA, Bernatchez L (2020) Using haplotype information for conservation genomics. Trends in Ecology & Evolution, 35, 245-258.
DOI URL |
| [13] |
Librado P, Rozas J (2009) DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics, 25, 1451-1452.
DOI PMID |
| [14] | Mastretta-Yanes A, da Silva JM, Grueber CE, Castillo-Reina L, Köppä V, Forester BR, Funk WC, Heuertz M, Ishihama F, Jordan R, Mergeay J, Paz-Vinas I, Rincon-Parra VJ, Rodriguez-Morales MA, Arredondo-Amezcua L, Brahy G, DeSaix M, Durkee L, Hamilton A, Hunter ME, Koontz A, Lang I, Latorre-Cárdenas MC, Latty T, Llanes-Quevedo A, MacDonald AJ, Mahoney M, Miller C, Ornelas JF, Ramírez-Barahona S, Robertson E, Russo IM, Santiago MA, Shaw RE, Shea GM, Sjögren-Gulve P, Spence ES, Stack T, Suárez S, Takenaka A, Thurfjell H, Turbek S, van der Merwe M, Visser F, Wegier A, Wood G, Zarza E, Laikre L, Hoban S (2024) Multinational evaluation of genetic diversity indicators for the Kunming-Montreal Global Biodiversity Framework. Ecology Letters, 27, e14461. |
| [15] |
Miraldo A, Li S, Borregaard MK, Flórez-Rodríguez A, Gopalakrishnan S, Rizvanovic M, Wang ZH, Rahbek C, Marske KA, Nogués-Bravo D (2016) An Anthropocene map of genetic diversity. Science, 353, 1532-1535.
PMID |
| [16] | Nei M, Li WH (1979) Mathematical model for studying genetic variation in terms of restriction endonucleases. Proceedings of the National Academy of Sciences, USA, 76, 5269-5273. |
| [17] |
Nei M, Roychoudhury AK (1974) Sampling variances of heterozygosity and genetic distance. Genetics, 76, 379-390.
DOI PMID |
| [18] |
Nei M, Tajima F (1981) DNA polymorphism detectable by restriction endonucleases. Genetics, 97, 145-163.
DOI PMID |
| [19] |
Romiguier J, Gayral P, Ballenghien M, Bernard A, Cahais V, Chenuil A, Chiari Y, Dernat R, Duret L, Faivre N, Loire E, Lourenco JM, Nabholz B, Roux C, Tsagkogeorga G, Weber AAT, Weinert LA, Belkhir K, Bierne N, Glémin S, Galtier N (2014) Comparative population genomics in animals uncovers the determinants of genetic diversity. Nature, 515, 261-263.
DOI |
| [1] | 贾晓旭, 陈皖强, 唐修君, 樊艳凤, 张静, 王海威, 高玉时. 西南地区家鸡线粒体DNA控制区遗传多样性和基因渗入[J]. 生物多样性, 2026, 34(5): 26003-. |
| [2] | 陈德付, 梁铭荣, 许益镌. 垂直空间层次蚂蚁采样方法概述[J]. 生物多样性, 2025, 33(8): 25078-. |
| [3] | 范平, 王欢, 温知新, 宋刚, 雷富民. 气候因子对鸟类遗传多样性与物种分布面积关系的影响[J]. 生物多样性, 2025, 33(8): 25072-. |
| [4] | 薛瑞翔, 马雪蓉, 吴炯文, 刘爱君, 张细权, 季从亮, 殷颖珊, 朱炜健, 罗庆斌. 中山麻鸭群体遗传多样性与遗传结构[J]. 生物多样性, 2025, 33(8): 24592-. |
| [5] | 范平, 温知新, 宋刚. 气候因子和人类活动对两栖及哺乳动物不同遗传多样性指标的影响[J]. 生物多样性, 2025, 33(8): 25022-. |
| [6] | 周智成, 曹天玲, 刘如垚, 丁琪琪, 马轲, 杨丽萍, 周传江, 聂国兴, 汤永涛. 基于线粒体COI基因的黄河流域麦穗鱼种群遗传多样性与遗传结构[J]. 生物多样性, 2025, 33(8): 24501-. |
| [7] | 王儒晓, 史博洋, 潘达, 孙红英. 中国特有华溪蟹属淡水蟹多样性格局及其保护空缺[J]. 生物多样性, 2025, 33(8): 25123-. |
| [8] | 洪德元. 分类学中的方法论小叙[J]. 生物多样性, 2025, 33(2): 24541-. |
| [9] | 张舒欣, 贾紫璇, 方涛, 刘一凡, 赵微, 王荣, 昌海超, 罗芳丽, 朱耀军, 于飞海. 植物抗逆能力评价方法研究进展[J]. 生物多样性, 2025, 33(2): 24168-. |
| [10] | 伍金山, 杨长乐, 马玉凤, 李亚旋, 高文家, 叶·库斯力, 叶樑洪, 杨宇骄, 徐梦琦, 廖廷琼, 钟林强, 单文娟. 艾比湖湿地国家级自然保护区马鹿遗传多样性及遗传结构[J]. 生物多样性, 2025, 33(12): 25233-. |
| [11] | 王嘉陈, 徐汤俊, 许唯, 张高季, 尤艺瑾, 阮宏华, 刘宏毅. 城市景观格局对大蚰蜒种群遗传结构的影响[J]. 生物多样性, 2025, 33(1): 24251-. |
| [12] | 谷际岐, 陈建平, 赖江山. 大语言模型在生物多样性研究中的应用[J]. 生物多样性, 2024, 32(9): 24258-. |
| [13] | 冯晨, 张洁, 黄宏文. 统筹植物就地保护与迁地保护的解决方案: 植物并地保护(parallel situ conservation)[J]. 生物多样性, 2023, 31(9): 23184-. |
| [14] | 李庆多, 栗冬梅. 全球蝙蝠巴尔通体流行状况分析[J]. 生物多样性, 2023, 31(9): 23166-. |
| [15] | 朱建国, 王林, 任国鹏. 《国家重点保护野生动物名录》调整的评估方法探讨[J]. 生物多样性, 2023, 31(8): 23045-. |
| 阅读次数 | ||||||
|
全文 |
|
|||||
|
摘要 |
|
|||||
备案号:京ICP备16067583号-7
Copyright © 2026 版权所有 《生物多样性》编辑部
地址: 北京香山南辛村20号, 邮编:100093
电话: 010-62836137, 62836665 E-mail: biodiversity@ibcas.ac.cn
![]()