
生物多样性 ›› 2026, Vol. 34 ›› Issue (1): 25369. DOI: 10.17520/biods.2025369 cstr: 32101.14.biods.2025369
所属专题: eDNA技术应用
闫姿伶1,2( ), 陈晓宇2(), 姚蒙1,2,*()(
), 陈晓宇2(), 姚蒙1,2,*()( )
)
收稿日期:2025-09-15
接受日期:2025-12-09
出版日期:2026-01-20
发布日期:2026-01-21
通讯作者:
姚蒙
基金资助:
Ziling Yan1,2(), Xiaoyu Chen2(), Meng Yao1,2,*()()
Received:2025-09-15
Accepted:2025-12-09
Online:2026-01-20
Published:2026-01-21
Contact:
Meng Yao
Supported by:摘要:
近年来, 环境DNA (eDNA)宏条形码技术被广泛应用于生物多样性研究, 但该技术在蓬勃发展的同时仍存在一些方法学问题有待解决。其中一个重要问题是生物信息学处理流程的选择, 尤其是对物种多样性极高的无脊椎动物, 测序结果的处理流程直接影响检测结果, 但目前缺乏对该过程的系统比较评估。本研究使用来源于淡水的eDNA样品进行无脊椎动物宏条形码测序, 比较评估多种生物信息学流程对于无脊椎动物序列处理的影响。研究中选取4种常用的聚类或降噪方法(UPARSE、Swarm、UNOISE和DADA2)以及3种分类分配方法(BOLDigger、BLASTN和朴素贝叶斯分类器), 共组合形成12种生物信息学处理流程。结果显示, DADA2降噪方法与BOLDigger分类分配相结合的处理流程产生了最多的无脊椎动物分子可操作分类单元(MOTU)与最高的分类覆盖度和分类分辨率。4种聚类或降噪方法中, UNOISE和DADA2降噪方法比UPARSE和Swarm聚类方法获得了更多的无脊椎动物MOTU; 3种分类分配方法中, BOLDigger和BLASTN比朴素贝叶斯分类器获得了更高的分类覆盖度和分类分辨率。这些结果对基于eDNA的淡水无脊椎动物多样性研究具有重要的参考价值, 此外还提示针对不同研究类群以及不同条形码区段, 需要相应调整使用的生物信息学方法, 以得到更为准确可靠的生物多样性数据。
闫姿伶, 陈晓宇, 姚蒙 (2026) 基于环境DNA宏条形码的无脊椎动物多样性研究: 生物信息学流程比较与评估. 生物多样性, 34, 25369. DOI: 10.17520/biods.2025369.
Ziling Yan, Xiaoyu Chen, Meng Yao (2026) A comparative evaluation of bioinformatic pipelines for invertebrate biodiversity profiling via environmental DNA metabarcoding. Biodiversity Science, 34, 25369. DOI: 10.17520/biods.2025369.
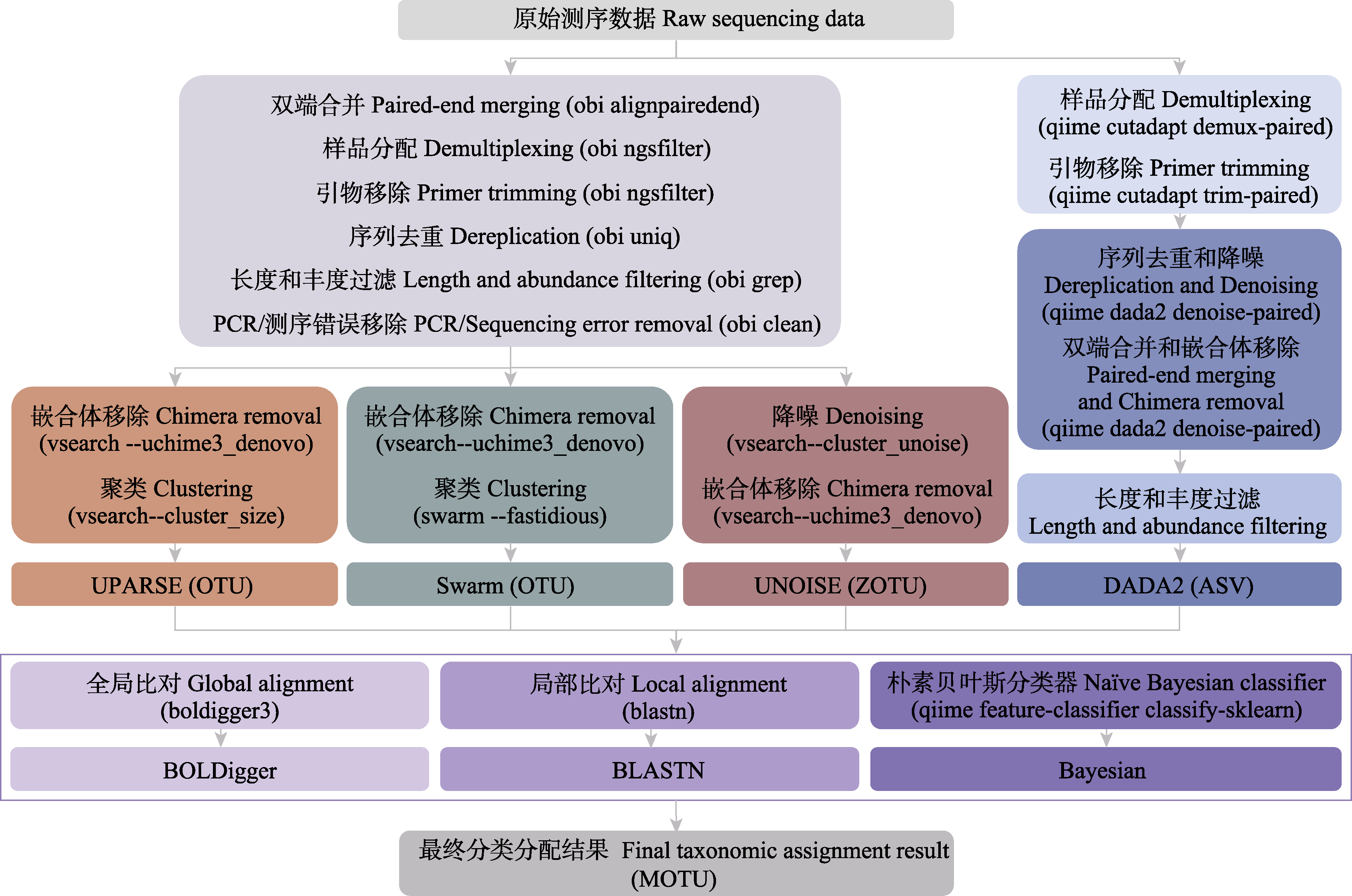
图1 本研究12种序列数据处理流程的具体步骤和命令
Fig. 1 The specific steps and commands of 12 pipelines used in this study
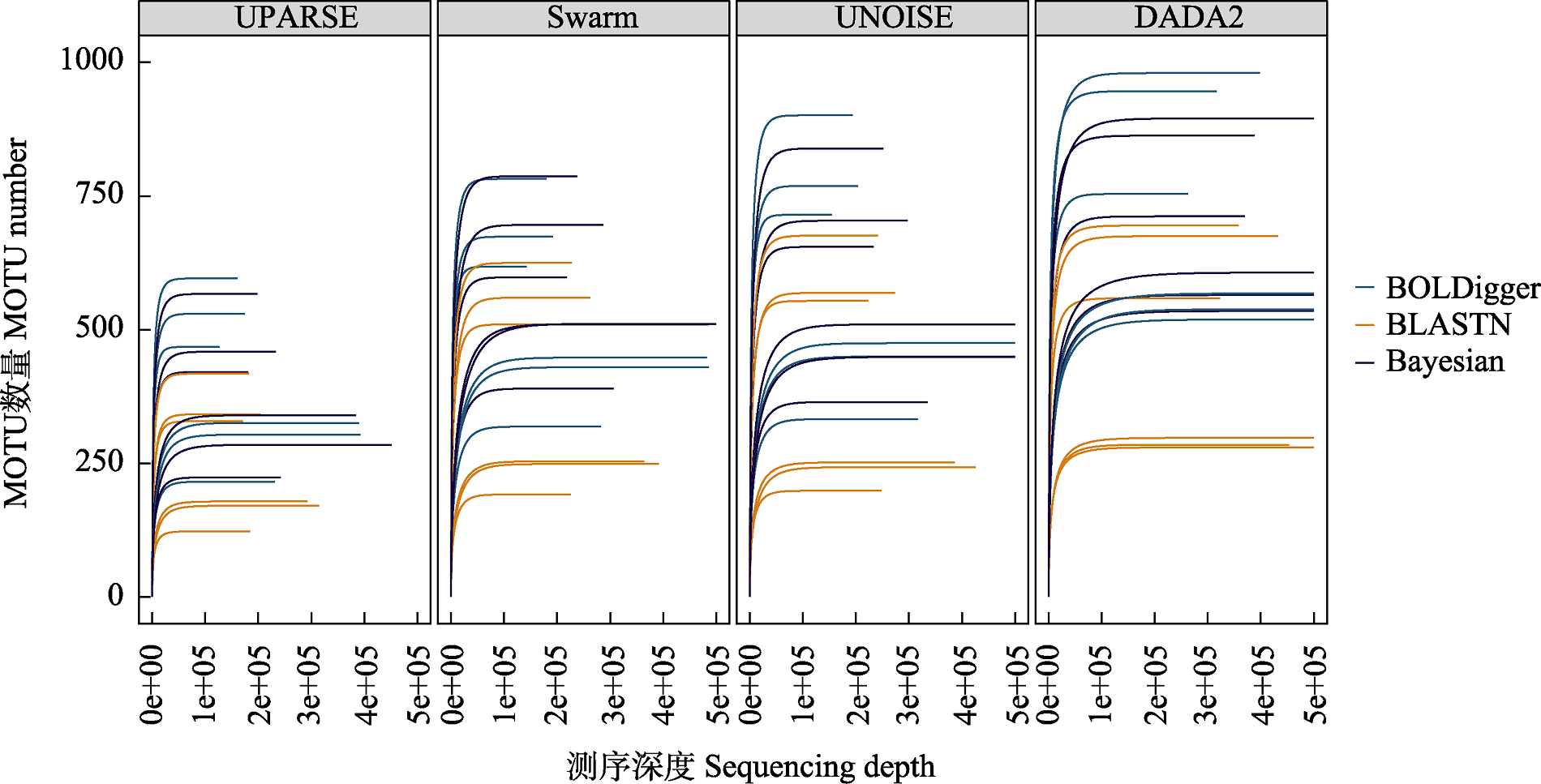
图2 不同处理流程检测出的无脊椎动物MOTU的稀疏曲线。每条曲线代表一个PCR产物。
Fig. 2 Rarefaction curve of invertebrate MOTU detected using different pipelines. Each curve represents a PCR product.
| 处理流程 Pipeline | MOTU (Reads) | 门 Phylum | 纲 Class | 目 Order | 科 Family | 属 Genus | 种 Species | |
|---|---|---|---|---|---|---|---|---|
| UPARSE | BOLDigger | 1,548 (1,475,851) | 13 | 27 | 71 | 141 | 108 | 111 |
| BLASTN | 984 (1,349,864) | 11 | 22 | 46 | 107 | 92 | 98 | |
| Bayesian | 1,391 (1,689,419) | 11 | 19 | 34 | 66 | 71 | 81 | |
| Swarm | BOLDigger | 2,142 (1,765,422) | 14 | 31 | 81 | 148 | 109 | 112 |
| BLASTN | 1,541 (1,680,656) | 11 | 22 | 47 | 108 | 94 | 104 | |
| Bayesian | 2,207 (2,095,227) | 11 | 19 | 34 | 67 | 72 | 82 | |
| UNOISE | BOLDigger | 2,392 (1,950,275) | 14 | 31 | 81 | 153 | 108 | 111 |
| BLASTN | 1,567 (1,797,455) | 11 | 22 | 47 | 108 | 94 | 104 | |
| Bayesian | 2,158 (2,264,348) | 11 | 19 | 34 | 67 | 73 | 82 | |
| DADA2 | BOLDigger | 2,777 (3,329,666) | 14 | 32 | 83 | 164 | 118 | 120 |
| BLASTN | 1,818 (2,678,329) | 11 | 23 | 49 | 113 | 102 | 108 | |
| Bayesian | 2,683 (3,690,943) | 11 | 19 | 35 | 71 | 78 | 87 | |
表1 12种处理流程获得的无脊椎动物MOTU和reads以及分类覆盖度。此处分类覆盖度指不同流程的产出结果在各分类水平上的覆盖广度。
Table 1 Invertebrate MOTU, reads and taxonomic coverage obtained from 12 pipelines. The taxonomic coverage here refers to the coverage breadth of the outputs from different pipelines across each taxonomic level.
| 处理流程 Pipeline | MOTU (Reads) | 门 Phylum | 纲 Class | 目 Order | 科 Family | 属 Genus | 种 Species | |
|---|---|---|---|---|---|---|---|---|
| UPARSE | BOLDigger | 1,548 (1,475,851) | 13 | 27 | 71 | 141 | 108 | 111 |
| BLASTN | 984 (1,349,864) | 11 | 22 | 46 | 107 | 92 | 98 | |
| Bayesian | 1,391 (1,689,419) | 11 | 19 | 34 | 66 | 71 | 81 | |
| Swarm | BOLDigger | 2,142 (1,765,422) | 14 | 31 | 81 | 148 | 109 | 112 |
| BLASTN | 1,541 (1,680,656) | 11 | 22 | 47 | 108 | 94 | 104 | |
| Bayesian | 2,207 (2,095,227) | 11 | 19 | 34 | 67 | 72 | 82 | |
| UNOISE | BOLDigger | 2,392 (1,950,275) | 14 | 31 | 81 | 153 | 108 | 111 |
| BLASTN | 1,567 (1,797,455) | 11 | 22 | 47 | 108 | 94 | 104 | |
| Bayesian | 2,158 (2,264,348) | 11 | 19 | 34 | 67 | 73 | 82 | |
| DADA2 | BOLDigger | 2,777 (3,329,666) | 14 | 32 | 83 | 164 | 118 | 120 |
| BLASTN | 1,818 (2,678,329) | 11 | 23 | 49 | 113 | 102 | 108 | |
| Bayesian | 2,683 (3,690,943) | 11 | 19 | 35 | 71 | 78 | 87 | |
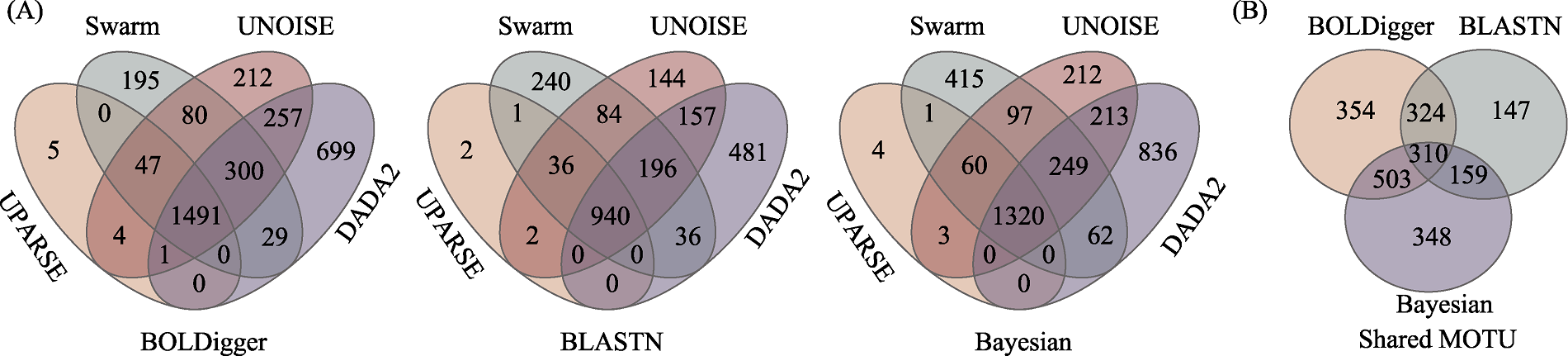
图3 不同聚类或降噪方法检测出的无脊椎动物MOTU数量(A)以及不同聚类或降噪方法共同检测出的MOTU在不同分类分配方法间比较(B)的韦恩图
Fig. 3 Venn diagrams of the number of invertebrate MOTU detected using different clustering or denoising methods (A) and the comparison of MOTU jointly detected using different clustering or denoising methods across different taxonomic assignment methods (B)
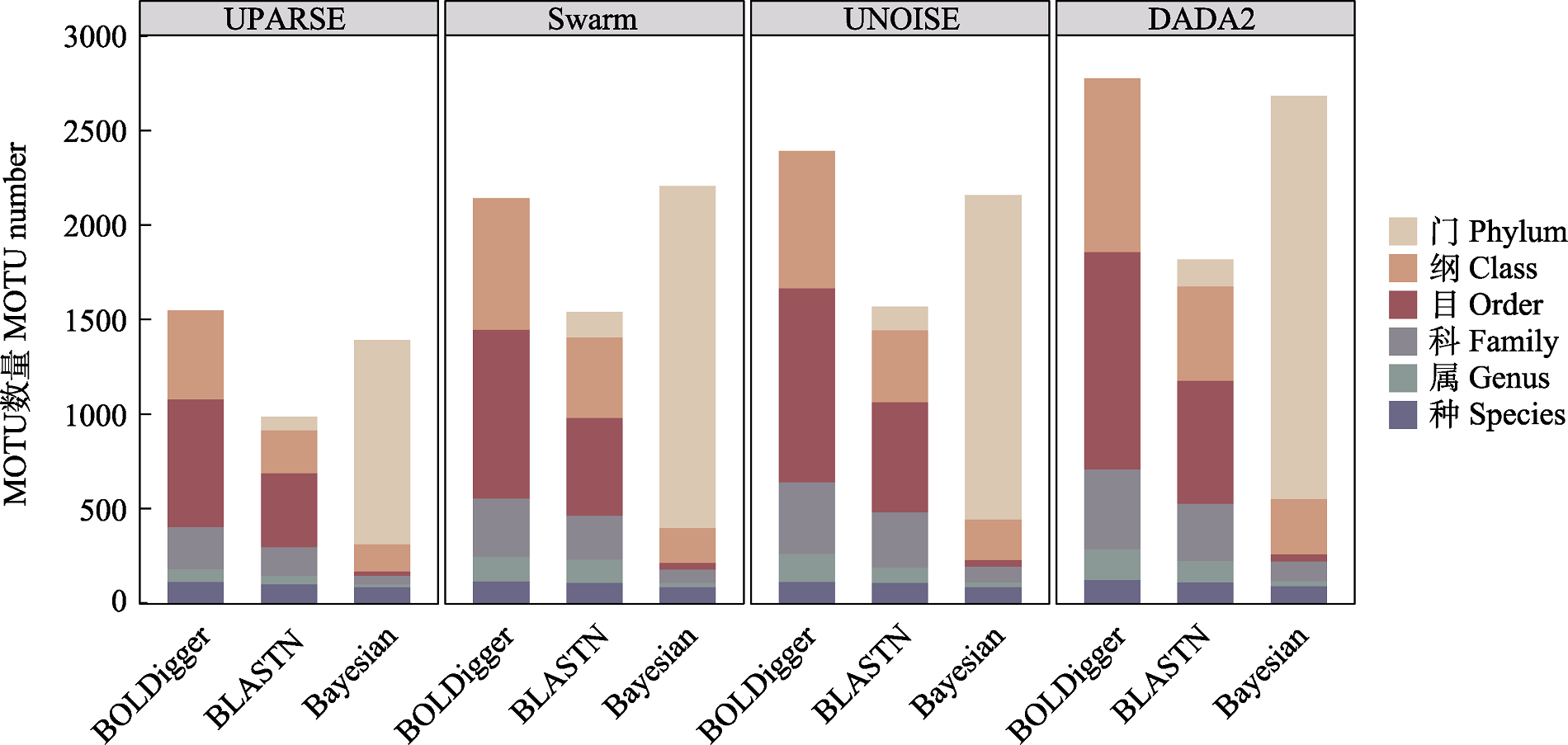
图4 不同处理流程检测出的无脊椎动物MOTU的分类分辨率。此处分类分辨率指不同流程鉴定结果的分类精度。
Fig. 4 The taxonomic resolution of invertebrate MOTU detected using different pipelines. The taxonomic resolution here refers to the taxonomic precision of the identification results from different pipelines.
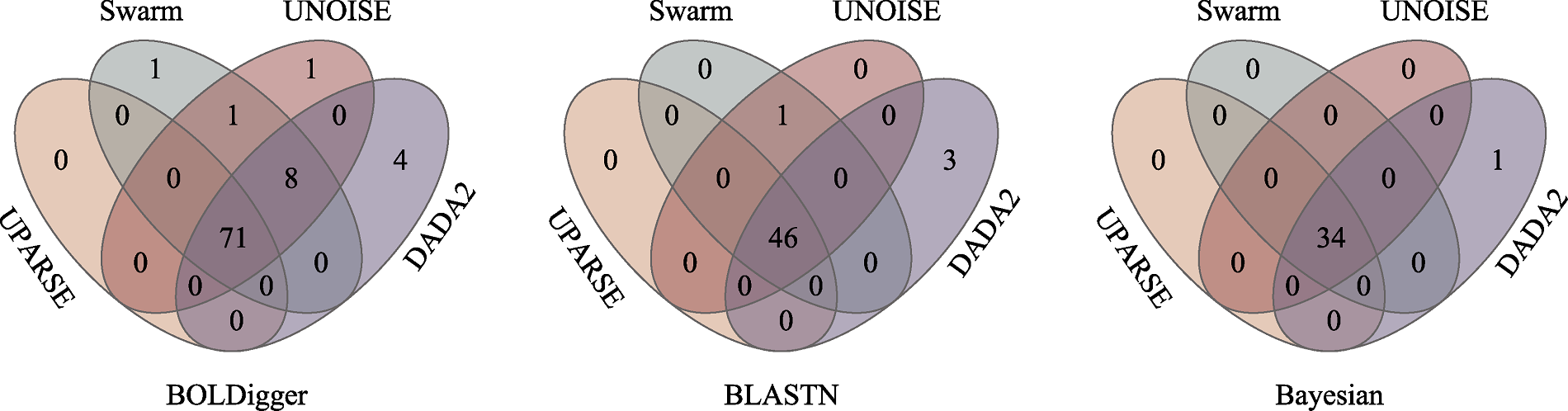
图5 不同聚类或降噪方法检测出的无脊椎动物目数量的韦恩图
Fig. 5 Venn diagrams of the number of invertebrate orders detected using different clustering or denoising methods
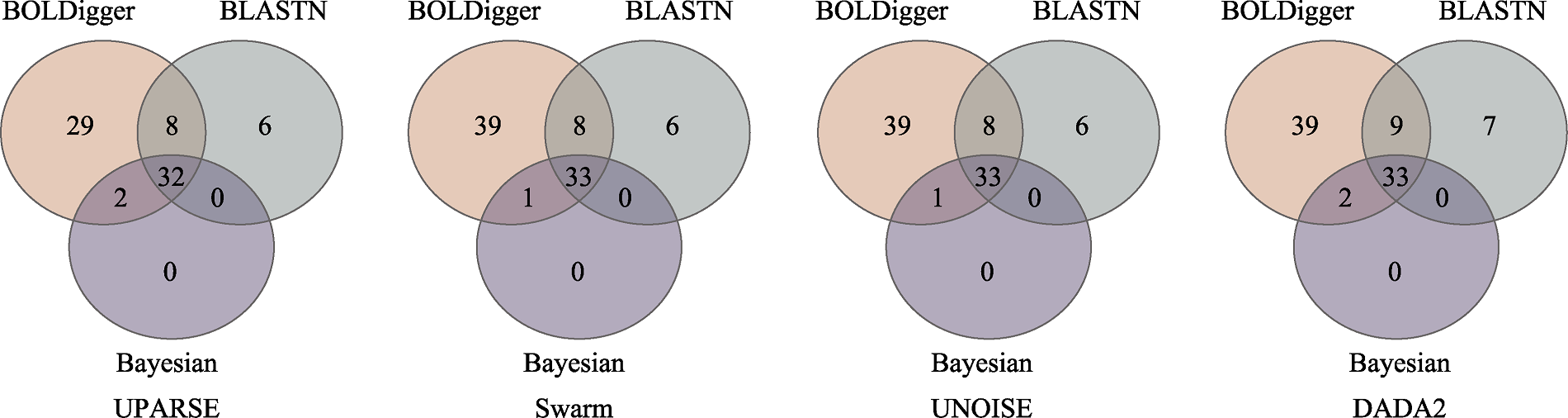
图6 不同分类分配方法检测出的无脊椎动物目数量的韦恩图
Fig. 6 Venn diagrams of the number of invertebrate orders detected using different taxonomic assignment methods
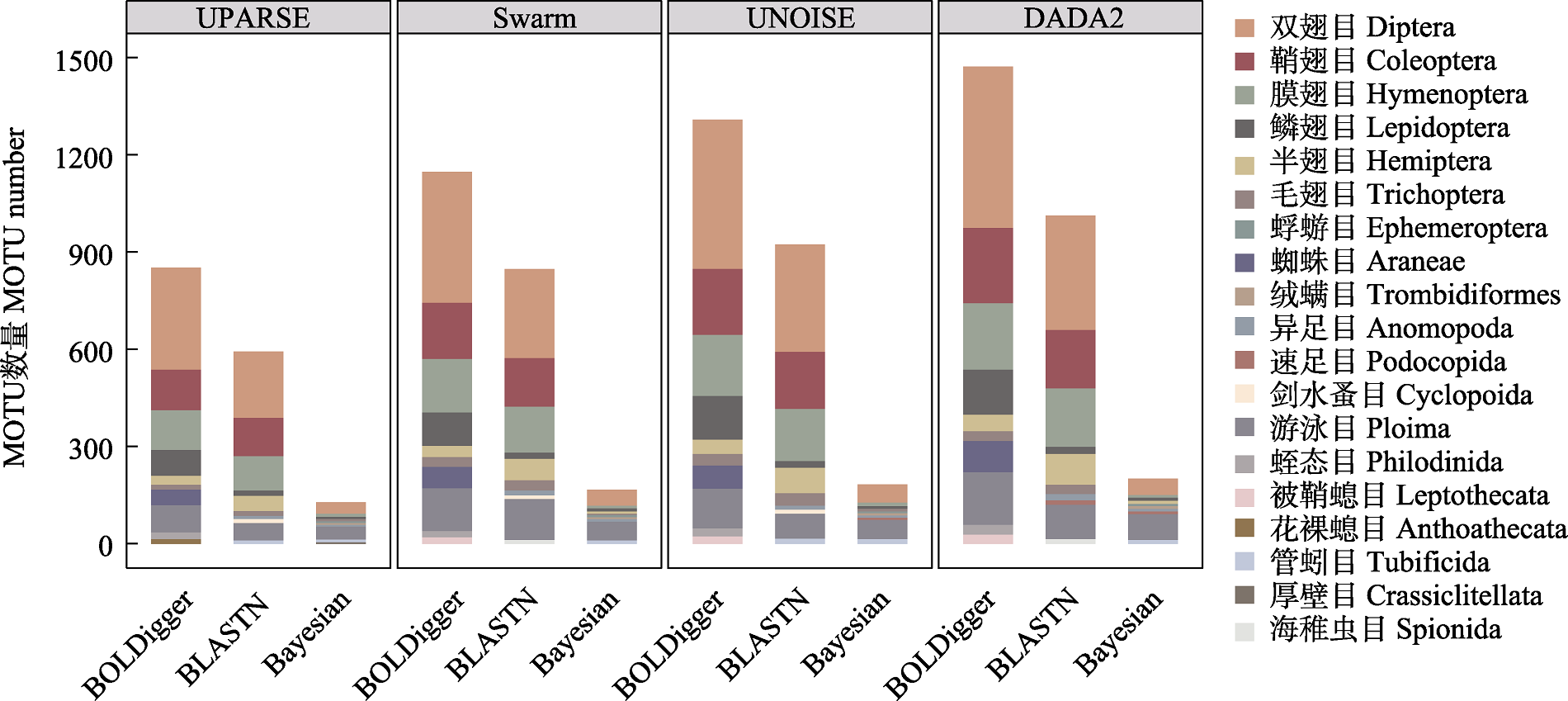
图7 不同处理流程检测出的属于主要无脊椎动物目的MOTU数量。仅显示各流程中MOTU数量排名前十位的目。
Fig. 7 The number of MOTU belong to major invertebrate orders detected using different pipelines. Only the top 10 orders with the largest number of MOTU are shown.
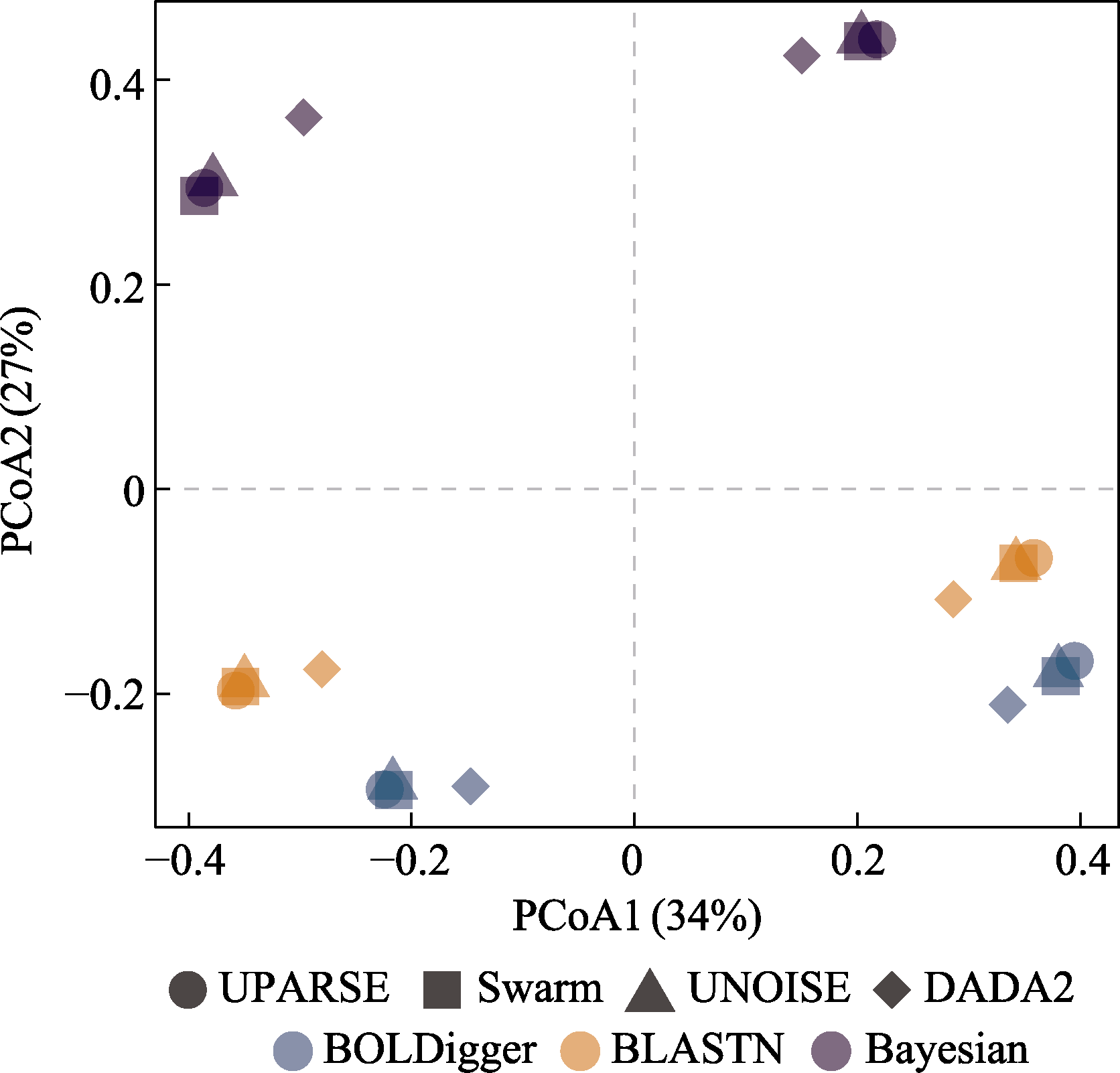
图8 基于Jaccard相异性指数对不同处理流程检测出的无脊椎动物MOTU进行的PCoA分析。形状和颜色均相同的符号对应河流和湖泊eDNA样品的结果。
Fig. 8 PCoA analysis of invertebrate MOTU detected using different pipelines based on Jaccard dissimilarity index. The symbols with the same shape and color correspond to the results of eDNA samples from rivers and lakes.
| [1] |
Alberdi A, Aizpurua O, Gilbert MTP, Bohmann K (2018) Scrutinizing key steps for reliable metabarcoding of environmental samples. Methods in Ecology and Evolution, 9, 134-147.
DOI URL |
| [2] |
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic Local Alignment Aearch Tool. Journal of Molecular Biology, 215, 403-410.
DOI PMID |
| [3] |
Antich A, Palacin C, Wangensteen OS, Turon X (2021) To denoise or to cluster, that is not the question: Optimizing pipelines for COI metabarcoding and metaphylogeography. BMC Bioinformatics, 22, 177.
DOI PMID |
| [4] | Bayer PE, Bennett A, Nester G, Corrigan S, Raes EJ, Cooper M, Ayad ME, McVey P, Kardailsky A, Pearce J, Fraser MW, Goncalves P, Burnell S, Rauschert S (2025) A comprehensive evaluation of taxonomic classifiers in marine vertebrate eDNA studies. Molecular Ecology Resources, 25, e14107. |
| [5] |
Bhat AH, Prabhu P, Balakrishnan K (2019) A critical analysis of state-of-the-art metagenomics OTU clustering algorithms. Journal of Biosciences, 44, 148.
DOI |
| [6] |
Bokulich NA, Kaehler BD, Rideout JR, Dillon M, Bolyen E, Knight R, Huttley GA, Caporaso JG (2018) Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome, 6, 90.
DOI |
| [7] | Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, Alexander H, Alm EJ, Arumugam M, Asnicar F, …, Warren J, Weber KC, Williamson CHD, Willis AD, Xu ZZ, Zaneveld JR, Zhang YL, Zhu QY, Knight R, Caporaso JG (2019) Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nature Biotechnology, 37, 852-857. |
| [8] |
Boyer F, Mercier C, Bonin A, Le Bras Y, Taberlet P, Coissac E (2016) Obitools: A unix-inspired software package for DNA metabarcoding. Molecular Ecology Resources, 16, 176-182.
DOI PMID |
| [9] |
Brandt MI, Trouche B, Quintric L, Günther B, Wincker P, Poulain J, Arnaud-Haond S (2021) Bioinformatic pipelines combining denoising and clustering tools allow for more comprehensive prokaryotic and eukaryotic metabarcoding. Molecular Ecology Resources, 21, 1904-1921.
DOI PMID |
| [10] | Buchner D, Leese F (2020) BOLDigger—A Python package to identify and organise sequences with the Barcode of Life Data systems. Metabarcoding and Metagenomics, 4, e53535. |
| [11] |
Bunholi IV, Foster NR, Casey JM (2023) Environmental DNA and RNA in aquatic community ecology: Toward methodological standardization. Environmental DNA, 5, 1133-1147.
DOI URL |
| [12] |
Cahyani NKD, Anggoro AW, Al Malik MD, Subhan B, Sani LMI, Madduppa H (2024) Inventorizing marine biodiversity using eDNA data from Indonesian coral reefs: Comparative high throughput analysis using different bioinformatic pipelines. Marine Biodiversity, 54, 39.
DOI |
| [13] |
Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP (2016) DADA2: High-resolution sample inference from Illumina amplicon data. Nature Methods, 13, 581-583.
DOI PMID |
| [14] |
Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL (2009) BLAST+: Architecture and applications. BMC Bioinformatics, 10, 421.
DOI PMID |
| [15] |
Chen XY, Yan ZL, Li S, Yao M (2025) Advancing aquatic biodiversity assessments of invertebrates using eDNA metabarcoding: A systematic evaluation of primers for marine and freshwater communities. Methods in Ecology and Evolution, 16, 2408-2430.
DOI URL |
| [16] |
Chernyshev AV, Kajihara H (2019) Comparative muscular morphology in archinemertea (Nemertea: Palaeonemertea). Zoomorphology, 138, 193-207.
DOI |
| [17] |
Coissac E (2012) OligoTag: A program for designing sets of tags for next-generation sequencing of multiplexed samples. Methods in Molecular Biology, 888, 13-31.
DOI PMID |
| [18] |
Cruaud P, Vigneron A, Fradette MS, Charette SJ, Rodriguez MJ, Dorea CC, Culley AI (2017) Open the SterivexTM casing: An easy and effective way to improve DNA extraction yields. Limnology and Oceanography: Methods, 15, 1015-1020.
DOI URL |
| [19] | De Grave S, Pentcheff ND, Ahyong ST, Chan TY, Crandall KA, Dworschak PC, Felder DL, Feldmann RM, Fransen CHJM, Goulding LYD, Lemaitre R, Low MEY, Martin JW, Ng PKL, Schweitzer CE, Tan SH, Tshudy D, Wetzer R (2009) A classification of living and fossil genera of decapod crustaceans. Raffles Bulletin of Zoology, 21, 1-109. |
| [20] |
De Santiago A, Pereira TJ, Mincks SL, Bik HM (2022) Dataset complexity impacts both MOTU delimitation and biodiversity estimates in eukaryotic 18S rRNA metabarcoding studies. Environmental DNA, 4, 363-384.
DOI URL |
| [21] |
Deiner K, Bik HM, Mächler E, Seymour M, Lacoursière- Roussel A, Altermatt F, Creer S, Bista I, Lodge DM, de Vere N, Pfrender ME, Bernatchez L (2017) Environmental DNA metabarcoding: Transforming how we survey animal and plant communities. Molecular Ecology, 26, 5872-5895.
DOI PMID |
| [22] |
Dumont HJ, Munuswamy N (1997) The potential of freshwater Anostraca for technical applications. Hydrobiologia, 358, 193-197.
DOI |
| [23] |
Edgar RC (2010) Search and clustering orders of magnitude faster than BLAST. Bioinformatics, 26, 2460-2461.
DOI PMID |
| [24] |
Edgar RC (2013) UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nature Methods, 10, 996-998.
DOI PMID |
| [25] | Edgar RC (2016) UNOISE2: Improved error-correction for Illumina 16S and ITS amplicon sequencing. bioRxiv preprint. https://doi.org/10.1101/081257. (accessed on 2025-07-13) |
| [26] | Eisenhauer N, Hines J (2021) Invertebrate biodiversity and conservation. Current Biology, 31, R1214-R1218. |
| [27] |
Folmer O, Black M, Hoeh W, Lutz R, Vrijenhoek R (1994) DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Molecular Marine Biology and Biotechnology, 3, 294-299.
PMID |
| [28] | Hakimzadeh A, Asbun AA, Albanese D, Bernard M, Buchner D, Callahan B, Caporaso JG, Curd E, Djemiel C, Durling MB, Elbrecht V, Gold Z, Gweon HS, Hajibabaei M, Hildebrand F, Mikryukov V, Normandeau E, Özkurt E, Palmer JM, Pascal G, Porter TM, Straub D, Vasar M, Větrovský T, Zafeiropoulos H, Anslan S (2024) A pile of pipelines: An overview of the bioinformatics software for metabarcoding data analyses. Molecular Ecology Resources, 24, e13847. |
| [29] |
Hao XL, Jiang R, Chen T (2011) Clustering 16S rRNA for OTU prediction: A method of unsupervised Bayesian clustering. Bioinformatics, 27, 611-618.
DOI PMID |
| [30] | Jackson MA, Bell JT, Spector TD, Steves CJ (2016) A heritability-based comparison of methods used to cluster 16S rRNA gene sequences into operational taxonomic units. PeerJ, 4, e2341. |
| [31] | Kopylova E, Navas-Molina JA, Mercier C, Xu ZZ, Mahé F, He Y, Zhou H-W, Rognes T, Caporaso JG, Knight R (2016) Open-source sequence clustering methods improve the state of the art. mSystems, 1, e00003-15. |
| [32] |
Leray M, Yang JY, Meyer CP, Mills SC, Agudelo N, Ranwez V, Boehm JT, Machida RJ (2013) A new versatile primer set targeting a short fragment of the mitochondrial COI region for metabarcoding metazoan diversity: Application for characterizing coral reef fish gut contents. Frontiers in Zoology, 10, 34.
DOI PMID |
| [33] | Li M, Wei TT, Shi BY, Hao XY, Xu HG, Sun HY (2019) Biodiversity monitoring of freshwater benthic macroinvertebrates using environmental DNA. Biodiversity Science, 27, 480-490. (in Chinese with English abstract) |
|
[李萌, 尉婷婷, 史博洋, 郝希阳, 徐海根, 孙红英 (2019) 环境DNA技术在淡水底栖大型无脊椎动物多样性监测中的应用. 生物多样性, 27, 480-490.]
DOI |
|
| [34] |
Li ZY, Zhao WC, Jiang Y, Wen YJ, Li M, Liu L, Zou KS (2024) New insights into biologic interpretation of bioinformatic pipelines for fish eDNA metabarcoding: A case study in Pearl River estuary. Journal of Environmental Management, 368, 122136.
DOI URL |
| [35] |
Lu Q, Zhang S-Y, Du JQ, Liu Q, Dong CX, Zhao JD, Wang YF, Yao M (2023) Multi-group biodiversity distributions and drivers of metacommunity organization along a glacial-fluvial-limnic pathway on the Tibetan Plateau. Environmental Research, 220, 115236.
DOI URL |
| [36] | Lynn DH (2008) The Ciliated Protozoa: Characterization, Classification, and Guide to the Literature, 3rd edn. Springer, New York. |
| [37] | Macé B, Hocdé R, Marques V, Guerin PE, Valentini A, Arnal V, Pellissier L, Manel S (2022) Evaluating bioinformatics pipelines for population-level inference using environmental DNA. Environmental DNA, 4, 674-686. |
| [38] |
Macher T-H, Beermann AJ, Arle J, Foerster J, Greyer M, Mora D, Koschorreck J, Rolauffs P, Rother A, Schüler S, Zimmermann J, Hering D, Leese F (2025) Fit for purpose? Evaluating benthic invertebrate DNA metabarcoding for ecological status class assessment in streams under the Water Framework Directive. Water Research, 272, 122987.
DOI URL |
| [39] |
Mahé F, Czech L, Stamatakis A, Quince C, de Vargas C, Dunthorn M, Rognes T (2021) Swarm v3: Towards tera-scale amplicon clustering. Bioinformatics, 38, 267-269.
DOI PMID |
| [40] |
Malica J, Rączka G, Turczański K, Andrzejewska A, Skorupski M, Urbanowski CK, Kamczyc J (2024) Impact of land use history and soil properties on soil mite communities (Acari, Mesostigmata) inhabiting stands growing on post-agricultural land. Land Degradation & Development, 35, 1776-1791.
DOI URL |
| [41] | Martin M (2011) Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet Journal, 17, 10-12. |
| [42] |
Mathon L, Valentini A, Guérin PE, Normandeau E, Noel C, Lionnet C, Boulanger E, Thuiller W, Bernatchez L, Mouillot D, Dejean T, Manel S (2021) Benchmarking bioinformatic tools for fast and accurate eDNA metabarcoding species identification. Molecular Ecology Resources, 21, 2565-2579.
DOI PMID |
| [43] | Mercier C, Boyer F, Bonin A, Coissac É (2013) SUMATRA and SUMACLUST: Fast and exact comparison and clustering of sequences. |
| [44] |
Múrria C, Wangensteen OS, Somma S, Väisänen L, Fortuño P, Arnedo MA, Prat N (2024) Taxonomic accuracy and complementarity between bulk and eDNA metabarcoding provides an alternative to morphology for biological assessment of freshwater macroinvertebrates. Science of the Total Environment, 935, 173243.
DOI URL |
| [45] | Nearing JT, Douglas GM, Comeau AM, Langille MGI (2018) Denoising the Denoisers: An independent evaluation of microbiome sequence error-correction approaches. PeerJ, 6, e5364. |
| [46] | Nilsen T, Snipen L-G, Angell IL, Keeley NB, Majaneva S, Pettersen R, Rudi K (2024) Swarm and UNOISE outperform DADA2 and Deblur for denoising high-diversity marine seafloor samples. ISME Communications, 4, ycae071. |
| [47] | Pawłowski J, Apothéloz-Perret-Gentil L, Mächler E, Altermatt F (2020) Apothéloz-Perret-Gentil L, Mächler E, Altermatt F (2020) Environmental DNA applications for biomonitoring and bioassessment in aquatic ecosystems. Guidelines. Federal Office for the Environment, Bern. Environmental Studies. no. 2010: 71 pp. https://doi.org/10.5167/uzh-187800. (accessed on 2025-07-26) |
| [48] |
Petit-Marty N, Casas L, Saborido-Rey F (2023) State-of-the-art of data analyses in environmental DNA approaches towards its applicability to sustainable fisheries management. Frontiers in Marine Science, 10, 1061530.
DOI URL |
| [49] | R Core Team (2023) R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria. https://www.R-project.org/. (accessed on 2025-09-04) |
| [50] | Ratnasingham S, Hebert PDN (2007) BOLD: The Barcode of Life Data System (http://www.barcodinglife.org). Molecular Ecology Notes, 7, 355-364. |
| [51] | Rogers DC (2013) Anostraca Catalogus (Crustacea: Branchiopoda). The Raffles Bulletin of Zoology, 61, 525-546. |
| [52] | Rognes T, Flouri T, Nichols B, Quince C, Mahé F (2016) VSEARCH: A versatile open source tool for metagenomics. PeerJ, 4, e2584. |
| [53] |
Rosen GL, Reichenberger ER, Rosenfeld AM (2011) NBC: The Naïve Bayes Classification tool webserver for taxonomic classification of metagenomic reads. Bioinformatics, 27, 127-129.
DOI PMID |
| [54] |
Sfecci E, Lacour T, Amade P, Mehiri M, Sfecci E, Lacour T, Amade P, Mehiri M (2016) Polycyclic guanidine alkaloids from Poecilosclerida marine sponges. Marine Drugs, 14, 77.
DOI URL |
| [55] | Tekle YI, Smith AR, McGinnis M, Ghebezadik S, Patel P (2025) A new Paramoeba isolate from Florida exhibits a microtubule-bound endosymbiont closely associated with the host nucleus. Journal of Eukaryotic Microbiology, 72, e70011. |
| [56] |
Turon X, Antich A, Palacín C, Præbel K, Wangensteen OS (2020) From metabarcoding to metaphylogeography: Separating the wheat from the chaff. Ecological Applications, 30, e02036.
DOI URL |
| [57] |
Vourka A, Karaouzas I, Parmakelis A (2023) River benthic macroinvertebrates and environmental DNA metabarcoding: A scoping review of eDNA sampling, extraction, amplification and sequencing methods. Biodiversity and Conservation, 32, 4221-4238.
DOI |
| [58] |
Wang Q, Garrity GM, Tiedje JM, Cole JR (2007) Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Applied and Environmental Microbiology, 73, 5261-5267.
DOI URL |
| [1] | 李云翱, 张文富, 赵桂刚, 杨春燕, 陈向清, 袁盛东, 曹敏, 蔡望, 杨洁. 空气环境DNA在陆生脊椎动物多样性监测上的应用: 以西双版纳20 ha森林动态样地为例[J]. 生物多样性, 2025, 33(6): 24318-. |
| [2] | 彭文, 邓泽帅, 郑文宝, 龚凌轩, 曾玉枫, 孟昊, 陈军, 杨道德. eDNA技术在两栖动物调查中的应用: 以湖南莽山国家级自然保护区为例[J]. 生物多样性, 2025, 33(6): 24552-. |
| [3] | 刘志禹, 吉鑫, 隋国辉, 杨定, 李轩昆. 北京首都国际机场野牛草与杂草草坪无脊椎动物多样性[J]. 生物多样性, 2025, 33(4): 24456-. |
| [4] | 尚华丹, 张楚晴, 王梅, 裴文娅, 李国宏, 王鸿斌. 中国杨树害虫物种多样性及其地理分布[J]. 生物多样性, 2025, 33(2): 24370-. |
| [5] | 寇毅秀, 翁朝红, 吉芬芬, 谢仰杰, 王家樵, 潘杭钊, 赵云廷, 叶坤. 环境DNA技术在濒危水生动物监测中的应用[J]. 生物多样性, 2025, 33(11): 24574-. |
| [6] | 姜熠辉, 刘岳, 曾旭, 林喆滢, 王楠, 彭吉豪, 曹玲, 曾聪. 东海六个国家级海洋保护区鱼类多样性和连通性[J]. 生物多样性, 2024, 32(6): 24128-. |
| [7] | 吴相獐, 雷富民, 单壹壹, 于晶. 上海城市公园苔藓植物多样性分布格局及其环境影响因子[J]. 生物多样性, 2024, 32(2): 23364-. |
| [8] | 董志远, 陈琳琳, 张乃鹏, 陈莉, 孙德斌, 倪艳梅, 李宝泉. 基于环境DNA宏条形码技术研究黄河三角洲典型潮沟系统鱼类多样性及其对水文连通性的响应[J]. 生物多样性, 2023, 31(7): 23073-. |
| [9] | 吴科毅, 阮文达, 周棣锋, 陈庆春, 张承云, 潘新园, 余上, 刘阳, 肖荣波. 基于音节聚类分析的被动声学监测技术及其在鸟类监测中的应用[J]. 生物多样性, 2023, 31(1): 22370-. |
| [10] | 刘丽平, 宋瑞凤, 张馥, 张秀香, 彭桂香, 谭志远. 高秆野生稻内生固氮细菌多样性[J]. 生物多样性, 2020, 28(8): 1018-1025. |
| [11] | 刘丹, 郭忠玲, 崔晓阳, 范春楠. 5种东北红豆杉植物群丛及其物种多样性的比较[J]. 生物多样性, 2020, 28(3): 340-349. |
| [12] | 李诣远, DavidC.Molik, MichaelE.Pfrender. |
| [13] | 刘山林. DNA条形码参考数据集构建和序列分析相关的新兴技术[J]. 生物多样性, 2019, 27(5): 526-533. |
| [14] | 王凤珍, 唐毅. 食物网关键种的判定及其对稳健性的影响[J]. 生物多样性, 2019, 27(10): 1132-1137. |
| [15] | 郜二虎, 何杰坤, 王志臣, 徐扬, 唐小平, 江海声. 全国陆生野生动物调查单元区划方案[J]. 生物多样性, 2017, 25(12): 1321-1330. |
| 阅读次数 | ||||||
|
全文 |
|
|||||
|
摘要 |
|
|||||
备案号:京ICP备16067583号-7
Copyright © 2026 版权所有 《生物多样性》编辑部
地址: 北京香山南辛村20号, 邮编:100093
电话: 010-62836137, 62836665 E-mail: biodiversity@ibcas.ac.cn
![]()