
Biodiv Sci ›› 2017, Vol. 25 ›› Issue (6): 577-599. DOI: 10.17520/biods.2017097 cstr: 32101.14.biods.2017097
Special Issue: 物种形成与系统进化
• Reviews • Previous Articles Next Articles
Jian-Feng Mao1,*( ), Yongpeng Ma2, Renchao Zhou3
), Yongpeng Ma2, Renchao Zhou3
Received:2017-03-26
Accepted:2017-05-04
Online:2017-06-20
Published:2017-07-10
Contact:
Mao Jian-Feng
Jian-Feng Mao, Yongpeng Ma, Renchao Zhou. Approaches used to detect and test hybridization: combining phylogenetic and population genetic analyses[J]. Biodiv Sci, 2017, 25(6): 577-599.
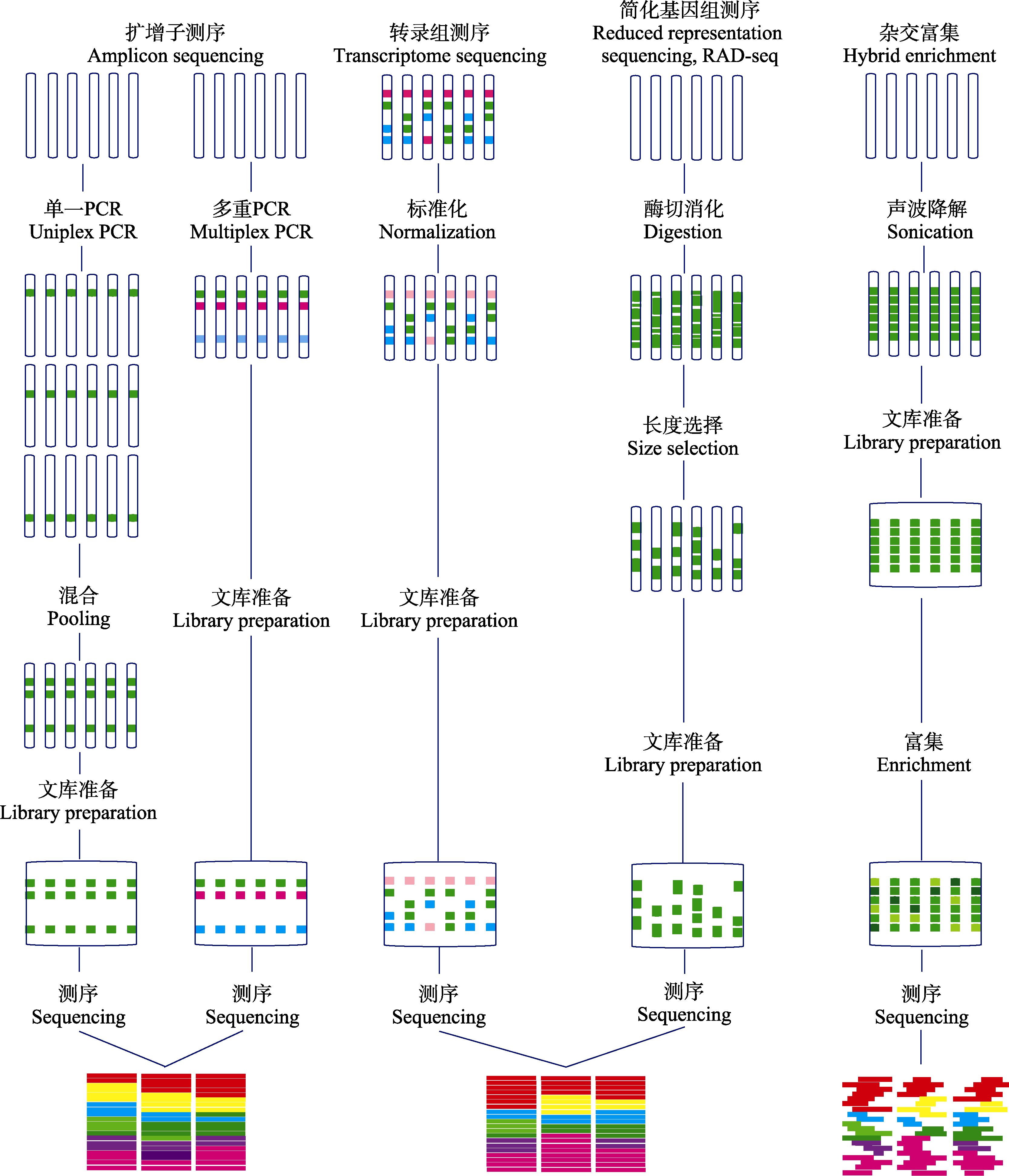
Fig. 1 Molecular genotyping technologies in the genomic era (adapted from Lemmon & Lemmon, 2013). The figure lists the features, operating procedures and data processing of each sequencing technique.
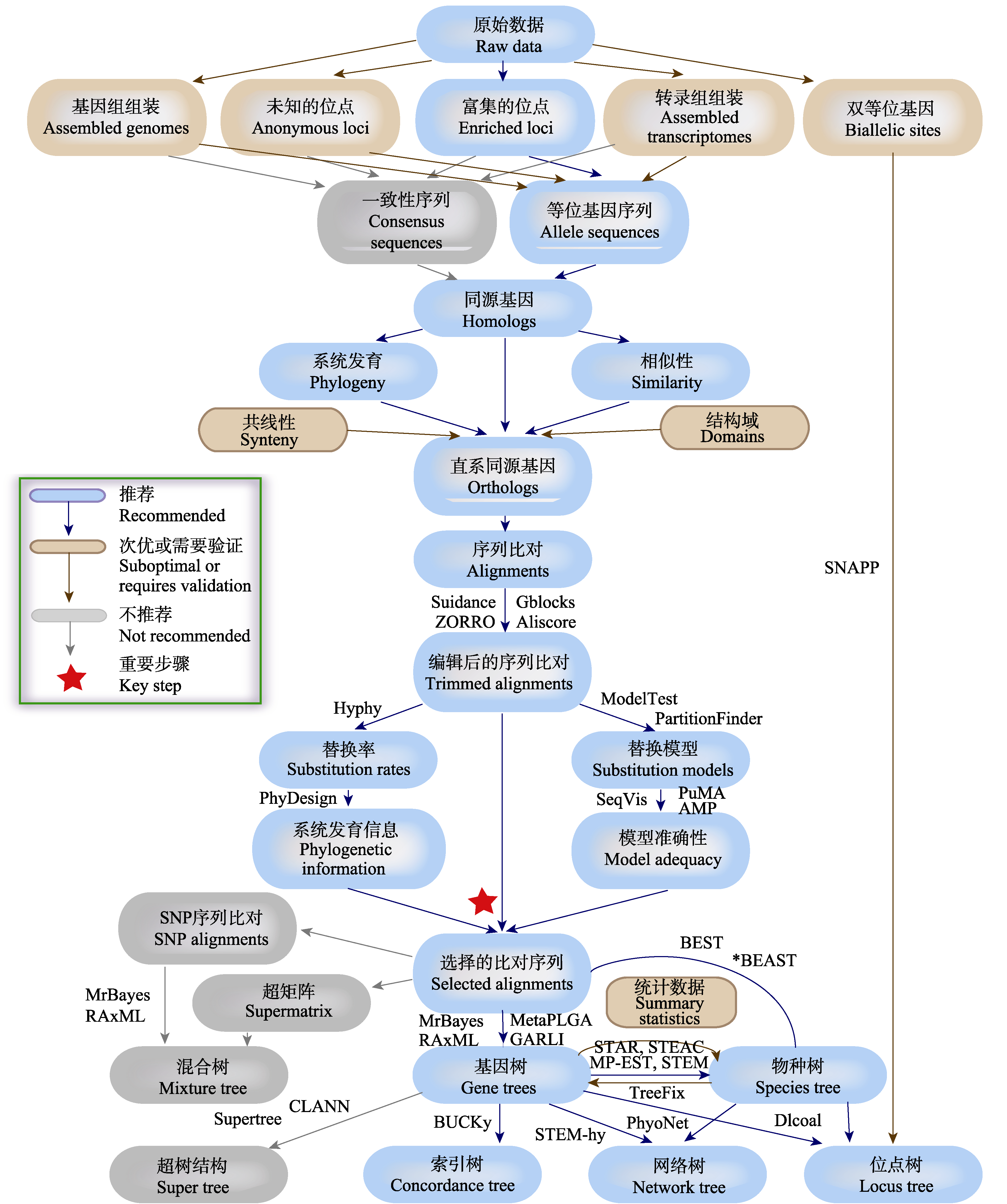
Fig. 2 The brief work-flow of phylogenetic strategy used to test hybridization (adapted from Lemmon & Lemmon, 2013). The work-flow contains multiple steps including data processing, data quality evaluation, data screening, orthologous gene identification, sequence analysis, selection of base substitution model, phylogenetic tree and phylogenetic network reconstruction. We sort out the hierarchy of recommended, suboptimal or requires validation and not recommended, in consideration of the degree of operation, the reliability of the data, whether can distinguish hybridization, incomplete lineage sorting and other factors. And we list the available softwares for each step.
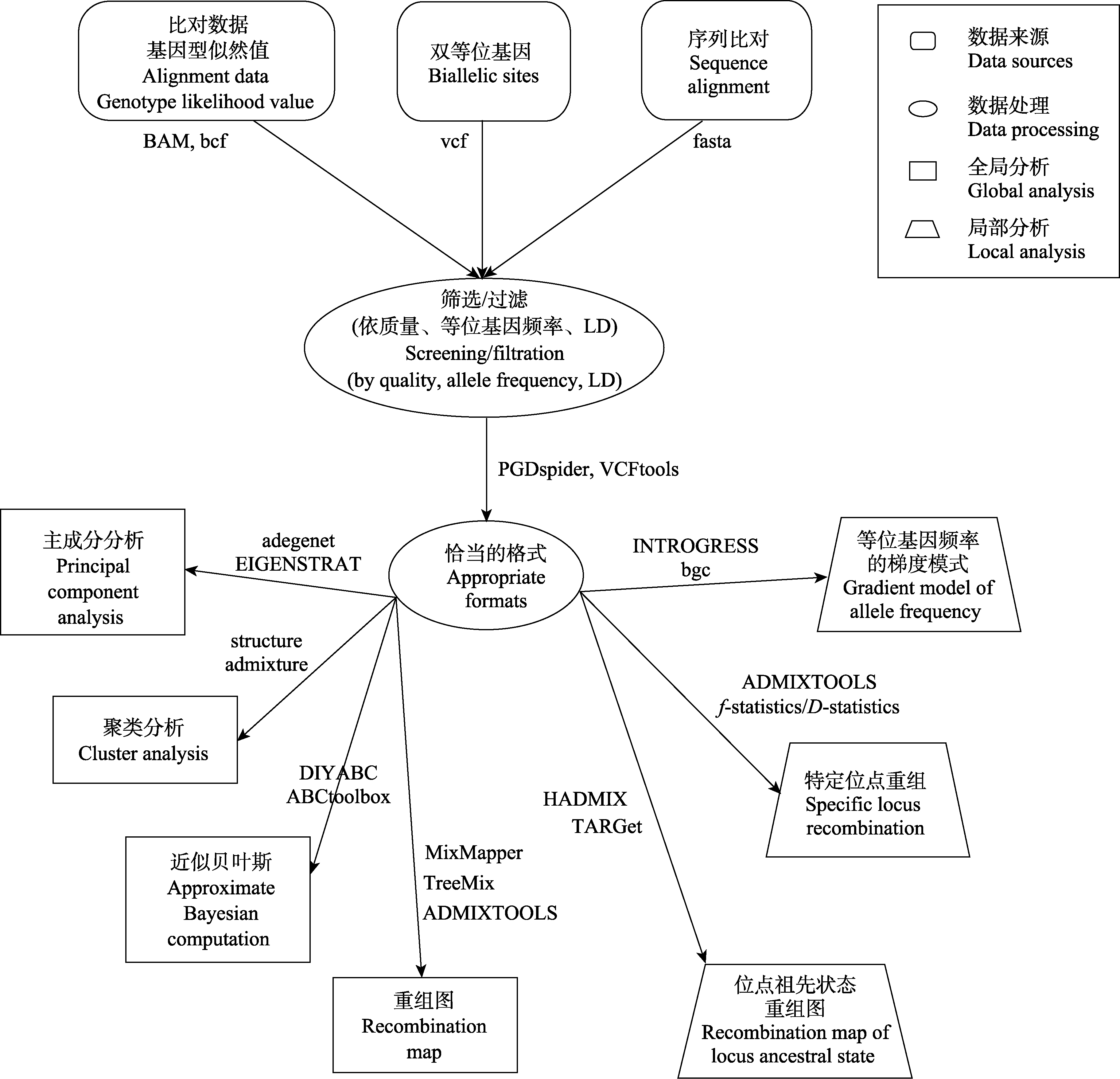
Fig. 3 The current population genomic work-flow used to test hybridization. The basic process of global and local testing for the existence and mode of hybridization through data collection, data processing and analysis are presented. The data format and some of the available softwares are listed.
| [1] | Abbott R, Albach D, Ansell S, Arntzen JW, Baird SJE, Bierne N, Boughman J, Brelsford A, Buerkle CA, Buggs R, Butlin RK, Dieckmann U, Eroukhmanoff F, Grill A, Cahan SH, Hermansen JS, Hewitt G, Hudson AG, Jiggins C, Jones J, Keller B, Marczewski T, Mallet J, Martinez-Rodriguez P, Möst M, Mullen S, Nichols R, Nolte AW, Parisod C, Pfennig K, Rice AM, Ritchie MG, Seifert B, Smadja CM, Stelkens R, Szymura JM, Väinölä R, Wolf JBW, Zinner D (2013) Hybridization and speciation. Journal of Evolutionary Biology, 26, 229-246. |
| [2] | Abby SS, Tannier E, Gouy M, Daubin V (2012) Lateral gene transfer as a support for the tree of life. Proceedings of the National Academy of Sciences, USA, 109, 4962-4967. |
| [3] | Ackermann RR, Mackay A, Arnold ML (2016) The hybrid origin of “Modern” humans. Evolutionary Biology, 43, 1-11. |
| [4] | Adams RP (1982) A comparison of multivariate methods for the detection of hybridization. Taxon, 31, 646-661. |
| [5] | Albert AYK, Schluter D (2005) Selection and the origin of species. Current Biology, 15, 283-288. |
| [6] | Albert TJ, Molla MN, Muzny DM, Nazareth L, Wheeler D, Song X, Richmond TA, Middle CM, Rodesch MJ, Packard CJ (2007) Direct selection of human genomic loci by microarray hybridization. Nature Methods, 4, 903-905. |
| [7] | Alexander DH, Novembre J, Lange K (2009) Fast model-based estimation of ancestry in unrelated individuals. Genome Research, 19, 1655-1664. |
| [8] | Anderson E, Hubricht L (1938) Hybridization in Tradescantia. III. The evidence for introgressive hybridization. American Journal of Botany, 25, 396-402. |
| [9] | Andrews KR, Good JM, Miller MR, Luikart G, Hohenlohe PA (2016) Harnessing the power of RADseq for ecological and evolutionary genomics. Nature Reviews Genetics, 17, 81-92. |
| [10] | Arnold ML (2016) Divergence with Genetic Exchange. Oxford University Press, Oxford. |
| [11] | Baird NA, Etter PD, Atwood TS, Currey MC, Shiver AL, Lewis ZA, Selker EU, Cresko WA, Johnson EA (2008) Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS ONE, 3, e3376. |
| [12] | Baran Y, Pasaniuc B, Sankararaman S, Torgerson DG, Gignoux C, Eng C, Rodriguezcintron W, Chapela R, Ford JG, Avila PC (2012) Fast and accurate inference of local ancestry in Latino populations. Bioinformatics, 28, 1359-1367. |
| [13] | Barton NH (1983) Multilocus clines. Evolution, 37, 454-471. |
| [14] | Bauer DC (2011) Variant calling comparison CASAVA1.8 and GATK. Nature Precedings, doi: 10.1038/npre.2011.6107.1/. |
| [15] | Beaumont MA (2010) Approximate Bayesian computation in evolution and ecology. Annual Review of Ecology, Evolution, and Systematics, 41, 379-406. |
| [16] | Bejerano G, Pheasant M, Makunin I, Stephen S, Kent WJ, Mattick JS, Haussler D (2004) Ultraconserved elements in the human genome. Science, 304, 1321-1325. |
| [17] | Boussau B, Szöllosi GJ, Duret L, Gouy M, Tannier E, Daubin V (2013) Genome-scale coestimation of species and gene trees. Genome Research, 23, 323-330. |
| [18] | Branstetter MG, Longino JT, Ward PS, Faircloth BC (2017) Enriching the ant tree of life: enhanced UCE bait set for genome-scale phylogenetics of ants and other Hymenoptera. Methods in Ecology and Evolution, 8, 768-776. |
| [19] | Brown R, Pasaniuc B (2014) Enhanced methods for local ancestry assignment in sequenced admixed individuals. PLoS Computational Biology, 10, e1003555. |
| [20] | Bujarski J (2013) Genetic recombination in plant-infecting messenger-sense RNA viruses: overview and research perspectives. Frontiers in Plant Science, 4, doi: 10.3389/fpls.2013.00068. |
| [21] | Burri R, Nater A, Kawakami T, Mugal CF, Olason PI, Smeds L, Suh A, Dutoit L, Bureš S, Garamszegi LZ (2015) Linked selection and recombination rate variation drive the evolution of the genomic landscape of differentiation across the speciation continuum of Ficedula flycatchers. Genome Research, 25, 1656-1665. |
| [22] | Cámara PG, Levine AJ, Rabadán R (2016) Inference of ancestral recombination graphs through topological data analysis. PLoS Computational Biology, 12, e1005071. |
| [23] | Castresana J (2000) Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Molecular Biology and Evolution, 17, 540-552. |
| [24] | Castric V, Bechsgaard J, Schierup MH, Vekemans X (2008) Repeated adaptive introgression at a gene under multiallelic balancing selection. PLoS Genetics, 4, e1000168. |
| [25] | Chan YC, Roos C, Inouemurayama M, Inoue E, Shih CC, Pei JC, Vigilant L (2010) Mitochondrial genome sequences effectively reveal the phylogeny of Hylobates gibbons. PLoS ONE, 5, e14419. |
| [26] | Chester M, Leitch AR, Soltis PS, Soltis DE (2010) Review of the application of modern cytogenetic methods (FISH/GISH) to the study of reticulation (polyploidy/hybridisation). Genes, 1, 166-192. |
| [27] | Choler P, Erschbamer B, Tribsch A, Gielly L, Taberlet P (2004) Genetic introgression as a potential to widen a species’ niche: insights from alpine Carex curvula. Proceedings of the National Academy of Sciences, USA, 101, 171-176. |
| [28] | Cohan FM (2002) Sexual isolation and speciation in bacteria. Genetica, 116, 359-370. |
| [29] | Cohan FM, Kane M (2001) Bacterial species and speciation. Systematic Biology, 50, 513-524. |
| [30] | Cornuet JM, Pudlo P, Veyssier J, Dehnegarcia A, Gautier M, Leblois R, Marin JM, Estoup A (2014) DIYABC v2.0: a software to make approximate Bayesian computation inferences about population history using single nucleotide polymorphism, DNA sequence and microsatellite data. Bioinformatics, 30, 1187. |
| [31] | Crawford NG, Faircloth BC, Mccormack JE, Brumfield RT, Winker K, Glenn TC (2012) More than 1000 ultraconserved elements provide evidence that turtles are the sister group of archosaurs. Biology Letters, 8, 783-786. |
| [32] | Csilléry K, Blum MGB, Gaggiotti OE, François O (2010) Approximate Bayesian computation (ABC) in practice. Trends in Ecology and Evolution, 25, 410-418. |
| [33] | Csilléry K, François O, Blum MGB (2011) abc: an R package for approximate Bayesian computation (ABC). Methods in Ecology and Evolution, 3, 475-479. |
| [34] | Davey JW, Hohenlohe PA, Etter PD, Boone JQ, Catchen JM, Blaxter ML (2011) Genome-wide genetic marker discovery and genotyping using next-generation sequencing. Nature Reviews Genetics, 12, 499-510. |
| [35] | Degnan JH, Rosenberg NA (2006) Discordance of species trees with their most likely gene trees. PLoS Genetics, 2, e68. |
| [36] | Degnan JH, Rosenberg NA (2009) Gene tree discordance, phylogenetic and the multispecies coalescent. Trends in Ecology and Evolution, 24, 332-340. |
| [37] | Dehal P, Boore JL (2005) Two rounds of whole genome duplication in the ancestral vertebrate. PLoS Biology, 3, e314. |
| [38] | Delport W, Poon AFY, Frost SDW, Pond SLK (2010) Datamonkey 2010: a suite of phylogenetic analysis tools for evolutionary biology. Bioinformatics, 26, 2455-2457. |
| [39] | Depotter JR, Seidl MF, Wood TA, Thomma BP (2016) Interspecific hybridization impacts host range and pathogenicity of filamentous microbes. Current Opinion in Microbiology, 32, 7-13. |
| [40] | DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, McKenna A, Fennell TJ, Kernytsky AM, Sivachenko AY, Cibulskis K, Gabriel SB, Altshuler D, Daly M (2011) A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nature Genetics, 43, 491-498. |
| [41] | der Sarkissian C, Ermini L, Schubert M, Yang MA, Librado P, Fumagalli M, Jónsson H, Bargal GK, Albrechtsen A, Vieira FG (2015) Evolutionary genomics and conservation of the endangered Przewalski’s horse. Current Biology, 25, 2577-2583. |
| [42] | Duncan KE, Istock CA, Graham JB, Ferguson N (1989) Genetic exchange between Bacillus subtilis and Bacillus licheniformis: variable hybrid stability and the nature of bacterial species. Evolution, 43, 1585-1609. |
| [43] | Durand EY, Patterson N, Reich D, Slatkin M (2011) Testing for ancient admixture between closely related populations. Molecular Biology and Evolution, 28, 2239-2252. |
| [44] | Earl AM, Losick R, Kolter R (2008) Ecology and genomics of Bacillus subtilis. Trends in Microbiology, 16, 269-275. |
| [45] | Ebersberger I, Strauss S, von Haeseler A (2009) HaMStR: profile hidden Markov model based search for orthologs in ESTs. BMC Evolutionary Biology, 9, doi:10.1186/1471-2148-9-157. |
| [46] | Edwards S (2016) Species trees. In: Encyclopedia of Evolutionary Biology (ed. Kliman R), pp. 236-244. Academic Press, Oxford. |
| [47] | Edwards SV, Liu L, Pearl DK (2007) High-resolution species trees without concatenation. Proceedings of the National Academy of Sciences, USA, 104, 5936-5941. |
| [48] | Ellstand NC, Elam D (1993) Population genetic consequences of small population size: implications for plant conservation. Annual Review of Ecology, Evolution, and Systematics, 24, 217-242. |
| [49] | Etter PD, Preston JL, Bassham S, Cresko WA, Johnson EA (2011) Local de novo assembly of RAD paired-end contigs using short sequencing reads. PLoS ONE, 6, e18561. |
| [50] | Falush D, Stephens M, Pritchard JK (2003) Inference of population sructure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics, 164, 1567-1587. |
| [51] | Felsenstein J (1978) Cases in which parsimony or compatibility methods will be positively misleading. Systematic Zoology, 27, 401-410. |
| [52] | Felsenstein J (2004) Inferring Phylogenies. Sinauer Associates, Sunderland. |
| [53] | Fitzpatrick BM (2013) Alternative forms for genomic clines. Ecology and Evolution, 3, 1951-1966. |
| [54] | Freeling M, Subramaniam S, Yano M, Tuberosa R (2009) Conserved noncoding sequences (CNSs) in higher plants. Current Opinion in Plant Biology, 12, 126-132. |
| [55] | Frichot E, Mathieu F, Trouillon T, Bouchard G, Francois O (2014) Fast and efficient estimation of individual ancestry coefficients. Genetics, 196, 973-983. |
| [56] | Fu QM, Li H, Moorjani P, Jay F, Slepchenko SM, Bondarev AA, Johnson PLF, Petri AA, Prüfer K, Filippo CD (2014) The genome sequence of a 45,000-year-old modern human from western Siberia. Nature, 514, 445-449. |
| [57] | Fu QM, Posth C, Hajdinjak M, Petr M, Mallick S, Fernandes D, Furtwängler A, Haak W, Meyer M, Mittnik A (2016) The genetic history of Ice Age Europe. Nature, 534, 200-205. |
| [58] | Gambette P, Berry V, Paul C (2012) Quartets and unrooted phylogenetic networks. Journal of Bioinformatics & Compututational Biology, 10, 1250004-1250023. |
| [59] | Gao H, Williamson S, Bustamante CD (2007) A Markov chain Monte Carlo approach for joint inference of population structure and inbreeding rates from multilocus genotype data. Genetics, 176, 1635-1651. |
| [60] | Gao J, Wang B, Mao JF, Ingvarsson P, Zeng QY, Wang XR (2012) Demography and speciation history of the homoploid hybrid pine Pinus densata on the Tibetan Plateau. Molecular Ecology, 21, 4811-4827. |
| [61] | Gnirke A, Melnikov A, Maguire J, Rogov P, Leproust EM, Brockman W, Fennell T, Giannoukos G, Fisher S, Russ C (2009) Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing. Nature Biotechnology, 27, 182-189. |
| [62] | Godden G, Jordon-Thaden I, Chamala S, Crowl AA, García N, Germain-Aubrey C, Heaney JM, Latvis M, Qi XS, Gitzendanner MA (2012) Making next-generation sequencing work for you: approaches and practical considerations for marker development and phylogenetics. Plant Ecology & Diversity, 5, 427-450. |
| [63] | Gompert Z, Buerkle CA (2012) bgc: software for Bayesian estimation of genomic clines. Molecular Ecology Resources, 12, 1168-1176. |
| [64] | Gompert Z, Buerkle CA (2009) A powerful regression-based method for admixture mapping of isolation across the genome of hybrids. Molecular Ecology, 18, 1207-1224. |
| [65] | Gompert Z, Buerkle CA (2010) INTROGRESS: a software package for mapping components of isolation in hybrids. Molecular Ecology Resources, 10, 378-384. |
| [66] | Gompert Z, Buerkle CA (2011) Bayesian estimation of genomic clines. Molecular Ecology, 20, 2111-2127. |
| [67] | Grant PR, Grant BR (2014) Evolutionary biology: speciation undone. Nature, 507, 178-179. |
| [68] | Green RE, Krause J, Briggs AW, Maricic T, Stenzel U, Kircher M, Patterson N, Li H, Zhai W, Fritz MH (2010) A draft sequence of the Neandertal genome. Science, 328, 710-722. |
| [69] | Griffin PC, Robin C, Hoffmann AA (2011) A next-generation sequencing method for overcoming the multiple gene copy problem in polyploid phylogenetics, applied to Poa grasses. BMC Biology, 9, 1-18. |
| [70] | Gross BL, Rieseberg LH (2005) The ecological genetics of homoploid hybrid speciation. Journal of Heredity, 96, 241-252. |
| [71] | Grunewald S, Spillner A, Bastkowski S, Bogershausen A (2013) SuperQ: computing supernetworks from quartets. Computational Biology & Bioinformatics IEEE/ACM Transactions, 10, 151-160. |
| [72] | Guillot G, Mortier F, Estoup A (2004) Geneland: a program for landscape genetics. Molecular Ecology Notes, 5, 712-715. |
| [73] | Guillot G, Estoup A, Mortier F, Cosson JF (2005) A spatial satistical model for landscape genetics. Genetics, 170, 1261-1280. |
| [74] | Gunnarsdóttir ED, Li M, Bauchet M, Finstermeier K, Stoneking M (2011) High-throughput sequencing of complete human mtDNA genomes from the Philippines. Genome Research, 21, 1-11. |
| [75] | Haak W, Lazaridis I, Patterson N, Rohland N, Mallick S, Llamas B, Brandt G, Nordenfelt S, Harney E, Stewardson K, Fu QM, Mittnik A, Banffy E, Economou C, Francken M, Friederich S, Pena RG, Hallgren F, Khartanovich V, Khokhlov A, Kunst M, Kuznetsov P, Meller H, Mochalov O, Moiseyev V, Nicklisch N, Pichler SL, Risch R, Rojo GMA, Roth C, Szecsenyi-Nagy A, Wahl J, Meyer M, Krause J, Brown D, Anthony D, Cooper A, Alt KW, Reich D (2015) Massive migration from the steppe was a source for Indo-European languages in Europe. Nature, 522, 207-211. |
| [76] | Haber M, Mezzavilla M, Xue YL, Comas D, Gasparini P, Zalloua P, Tyler-Smith C (2016) Genetic evidence for an origin of the Armenians from Bronze Age mixing of multiple populations. European Journal of Human Genetics, 24, 931-936. |
| [77] | Hapke A, Thiele D (2016) GIbPSs: a toolkit for fast and accurate analyses of genotyping-by-sequencing data without a reference genome. Molecular Ecology Resources, 16, 979-990. |
| [78] | Hegarty MJ, Hiscock SJ (2008) Genomic clues to the evolutionary success of polyploid plants. Current Biology, 18, R435-R444. |
| [79] | Heled J, Drummond AJ (2010) Bayesian inference of species trees from multilocus data. Molecular Biology and Evolution, 27, 570-580. |
| [80] | Hellenthal G, Busby GBJ, Band G, Wilson JF, Capelli C, Falush D, Myers S (2014) A genetic atlas of human admixture history. Science, 343, 747-751. |
| [81] | Hodges E, Xuan Z, Balija V, Kramer M, Molla MN, Smith SW, Middle CM, Rodesch MJ, Albert TJ, Hannon GJ (2007) Genome-wide in situ exon capture for selective resequencing. Nature Genetics, 39, 1522-1527. |
| [82] | Huang CH, Sun RR, Hu Y, Zeng LP, Zhang N, Cai LM, Zhang Q, Koch MA, Ihsan AS, Edger PP (2015) Resolution of Brassicaceae phylogeny using nuclear genes uncovers nested radiations and supports convergent morphological evolution. Molecular Biology and Evolution, 33, 394-412. |
| [83] | Huson DH, Scornavacca C (2011) A survey of combinatorial methods for phylogenetic networks. Genome Biology and Evolution, 3, 23-35. |
| [84] | Jombart T (2008) adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics, 24, 1403-1405. |
| [85] | Jones G, Sagitov S, Oxelman B (2013) Statistical inference of allopolyploid species networks in the presence of incomplete lineage sorting. Systematic Biology, 62, 467-478. |
| [86] | Kenny EM (2011) Multiplex target enrichment using DNA indexing for ultra-high throughput SNP Detection. DNA Research, 18, 31-38. |
| [87] | Kim C, Guo H, Kong WQ, Chandnani R, Shuang LS, Paterson AH (2016) Application of genotyping by sequencing technology to a variety of crop breeding programs. Plant Science, 242, 14-22. |
| [88] | Kim ES, Rothschild MF (2014) Genomic adaptation of admixed dairy cattle in East Africa. Frontiers in Genetics, 5, doi: 10.3389/fgene.2014.00443. |
| [89] | Kim M, Cui ML, Cubas P, Gillies A, Lee K, Chapman MA, Abbott RJ, Coen E (2008) Regulatory genes control a key morphological and ecological trait transferred between species. Science, 322, 1116-1119. |
| [90] | Kosakovsky PSL, Posada D, Gravenor MB, Woelk CH, Frost SDW (2006) GARD: a genetic algorithm for recombination detection. Bioinformatics, 22, 3096-3098. |
| [91] | Kronforst MR, Hansen MEB, Crawford NG, Gallant JR, Zhang W, Kulathinal RJ, Kapan DD, Mullen SP (2013) Hybridization reveals the evolving genomic architecture of speciation. Cell Reports, 5, 666-677. |
| [92] | Kubatko LS, Carstens BC, Knowles LL (2009) STEM: species tree estimation using maximum likelihood for gene trees under coalescence. Bioinformatics, 25, 971-973. |
| [93] | Kubatko LS, Degnan JH (2007) Inconsistency of phylogenetic estimates from concatenated data under coalescence. Systematic Biology, 56, 17-24. |
| [94] | Kumar S (2012) Statistics and truth in phylogenomics. Molecular Biology and Evolution, 29, 457-472. |
| [95] | Lanfear R, Calcott B, Ho SY, Guindon S (2012) Partitionfinder: combined selection of partitioning schemes and substitution models for phylogenetic analyses. Molecular Biology and Evolution, 29, 1695-1701. |
| [96] | Lazaridis I, Patterson N, Mittnik A, Renaud G, Mallick S, Kirsanow K, Sudmant PH, Schraiber JG, Castellano S, Lipson M, Berger B, Economou C, Bollongino R, Fu QM, Bos KI, Nordenfelt S, Li H, de Filippo C, Prufer K, Sawyer S, Posth C, Haak W, Hallgren F, Fornander E, Rohland N, Delsate D, Francken M, Guinet JM, Wahl J, Ayodo G, Babiker HA, Bailliet G, Balanovska E, Balanovsky O, Barrantes R, Bedoya G, Ben-Ami H, Bene J, Berrada F, Bravi CM, Brisighelli F, Busby GBJ, Cali F, Churnosov M, Cole DEC, Corach D, Damba L, van Driem G, Dryomov S, Dugoujon JM, Fedorova SA, Gallego RI, Gubina M, Hammer M, Henn BM, Hervig T, Hodoglugil U, Jha AR, Karachanak-Yankova S, Khusainova R, Khusnutdinova E, Kittles R, Kivisild T, Klitz W, Kucinskas V, Kushniarevich A, Laredj L, Litvinov S, Loukidis T, Mahley RW, Melegh B, Metspalu E, Molina J, Mountain J, Nakkalajarvi K, Nesheva D, Nyambo T, Osipova L, Parik J, Platonov F, Posukh O, Romano V, Rothhammer F, Rudan I, Ruizbakiev R, Sahakyan H, Sajantila A, Salas A, Starikovskaya EB, Tarekegn A, Toncheva D, Turdikulova S, Uktveryte I, Utevska O, Vasquez R, Villena M, Voevoda M, Winkler CA, Yepiskoposyan L, Zalloua P, Zemunik T, Cooper A, Capelli C, Thomas MG, Ruiz-Linares A, Tishkoff SA, Singh L, Thangaraj K, Villems R, Comas D, Sukernik R, Metspalu M, Meyer M, Eichler EE, Burger J, Slatkin M, Paabo S, Kelso J, Reich D, Krause J (2014) Ancient human genomes suggest three ancestral populations for present-day Europeans. Nature, 513, 409-413. |
| [97] | Leaché AD, Rannala B (2010) The accuracy of species tree estimation under simulation: a comparison of methods. Systematic Biology, 60, 126-137. |
| [98] | Lefeuvre P, Moriones E (2015) Recombination as a motor of host switches and virus emergence: geminiviruses as case studies. Current Opinion in Virology, 10, 14-19. |
| [99] | Lemmon AR, Brown JM, Stanger-Hall K, Lemmon EM (2009) The effect of ambiguous data on phylogenetic estimates obtained by maximum likelihood and Bayesian inference. Systematic Biology, 58, 130-145. |
| [100] | Lemmon AR, Moriarty EC (2004) The importance of proper model assumption in Bayesian phylogenetics. Systematic Biology, 53, 265-277. |
| [101] | Lemmon EM, Lemmon AR (2013) High-throughput genomic data in systematics and phylogenetics. Annual Review of Ecology, Evolution, and Systematics, 44, 99-121. |
| [102] | Liang D, Mao JF, Zhao W, Zhou XQ, Yuan HW, Wang LM, Xing FQ, Wang XR, Li Y (2013) Seedling performance of Pinus densata and its parental population in the habitat of P. tabuliformis. Chinese Journal of Plant Ecology, 37, 150-163. (in Chinese with English abstract) |
| [梁冬, 毛建丰, 赵伟, 周先清, 袁虎威, 王黎明, 邢芳倩, 王晓茹, 李悦 (2013) 高山松及其亲本种群在油松生境下的苗期性状. 植物生态学报, 37, 150-163.] | |
| [103] | Lintusaari J, Gutmann MU, Dutta R, Kaski S, Corander J (2016) Fundamentals and recent developments in approximate Bayesian computation. Systematic Biology, 66, 66-82. |
| [104] | Lipson M, Loh PR, Levin A, Reich D, Patterson N, Berger B (2013) Efficient moment-based inference of admixture parameters and sources of gene flow. Molecular Biology and Evolution, 30, 1788-1802. |
| [105] | Lipson M, Loh PR, Patterson N, Moorjani P, Ko YC, Stoneking M, Berger B, Reich D (2014) Reconstructing Austronesian population history in Island Southeast Asia. Nature Communications, 5, doi: 10.1038/ncomms5689. |
| [106] | Liu L (2008) BEST: Bayesian estimation of species trees under the coalescent model. Bioinformatics, 24, 2542-2543. |
| [107] | Liu LQ, Gu ZJ (2011) Genomic in situ hybridization identifies genome donors of Camellia reticulata (Theaceae). Plant Science, 180, 554-559. |
| [108] | Liu L, Yu LL (2011) Estimating species trees from unrooted gene trees. Systematic Biology, 60, 661-667. |
| [109] | Liu L, Yu LL, Edwards SV (2010) A maximum pseudo-likelihood approach for estimating species trees under the coalescent model. BMC Evolutionary Biology, 10, doi: 10.1186/1471-2148-10-302. |
| [110] | Livak KJ (2003) SNP genotyping by the 5’-nuclease reaction. Methods in Molecular Biology, 212, 129-147. |
| [111] | Mörseburg A, Pagani L, Ricaut FX, Yngvadottir B, Harney E, Castillo C, Hoogervorst T, Antao T, Kusuma P, Brucato N (2016) Multi-layered population structure in Island Southeast Asians. European Journal of Human Genetics, 24, 1605-1611. |
| [112] | Ma XF, Szmidt AE, Wang XR (2006) Genetic structure and evolutionary history of a diploid hybrid pine Pinus densata inferred from the nucleotide variation at seven gene loci. Molecular Biology and Evolution, 23, 807-816. |
| [113] | Mallet J (2005) Hybridization as an invasion of the genome. Trends in Ecology and Evolution, 20, 229-237. |
| [114] | Mallet J, Besansky N, Hahn MW (2016) How reticulated are species? BioEssays, 38, 140-149. |
| [115] | Mamanova L, Coffey AJ, Scott CE, Kozarewa I, Turner EH, Kumar A, Howard E, Shendure J, Turner DJ (2010) Target-enrichment strategies for next-generation sequencing. Nature Methods, 7, 111-118. |
| [116] | Mao JF, Li Y, Wang XR (2009) Empirical assessment of the reproductive fitness components of the hybrid pine Pinus densata on the Tibetan Plateau. Evolutionary Ecology, 23, 447-462. |
| [117] | Maples BK, Gravel S, Kenny EE, Bustamante CD (2013) RFMix: a discriminative modeling approach for rapid and robust local-ancestry inference. American Journal of Human Genetics, 93, 278-288. |
| [118] | Marczewski T, Ma YP, Zhang XM, Sun WB, Marczewski AJ (2016) Why is population information crucial for taxonomy? A case study involving a hybrid swarm and related varieties. AoB Plants, 8, doi:10.1093/aobpla/plw070. |
| [119] | Maricic T, Whitten M, Pääbo S (2010) Multiplexed DNA sequence capture of mitochondrial genomes using PCR products. PLoS ONE, 5, e14004. |
| [120] | Marioni JC, Mason CE, Mane SM, Stephens M, Gilad Y (2008) RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome Research, 18, 1509-1517. |
| [121] | Martin DP, Murrell B, Golden M, Khoosal A, Muhire B (2015) RDP4: detection and analysis of recombination patterns in virus genomes. Chemical Research in Toxicology, 1, doi: 10.1093/ve/vev003. |
| [122] | Martin JA, Wang Z (2011) Next-generation transcriptome assembly. Nature Reviews Genetics, 12, 671-682. |
| [123] | Martin NH, Bouck AC, Arnold ML (2006) Detecting adaptive trait introgression between Iris fulva and I. brevicaulis in highly selective field conditions. Genetics, 172, 2481-2489. |
| [124] | Martinsen GD, Whitham TG, Turek RJ, Keim P (2001) Hybrid populations selectively filter gene introgression between species. Evolution, 55, 1325-1335. |
| [125] | Mayer C, Sann M, Donath A, Meixner M, Podsiadlowski L, Peters RS, Petersen M, Meusemann K, Liere K, Wägele JW, Misof B, Bleidorn C, Ohl M, Niehuis O (2016) BaitFisher: a software package for multispecies target DNA enrichment probe design. Molecular Biology and Evolution, 33, 1875-1886. |
| [126] | Mccormack JE, Al E (2011) Next-generation sequencing reveals phylogeographic structure and a species tree for recent bird divergences. Molecular Phylogenetics & Evolution, 62, 397-406. |
| [127] | Mcguire G, Wright F, Prentice MJ (1997) A graphical method for detecting recombination in phylogenetic data sets. Molecular Biology and Evolution, 14, 1125-1131. |
| [128] | McKinney GJ, Waples RK, Seeb LW, Seeb JE (2016) Paralogs are revealed by proportion of heterozygotes and deviations in read ratios in genotyping-by-sequencing data from natural populations. Molecular Ecology Resources, 17, 656-669. |
| [129] | Mcvean G (2009) A genealogical interpretation of principal components analysis. PLoS Genetics, 5, e1000686. |
| [130] | Meier JI, Marques DA, Mwaiko S, Wagner CE, Excoffier L, Seehausen O (2017) Ancient hybridization fuels rapid cichlid fish adaptive radiations. Nature Communications, 8, doi: 10.1038/ncomms14363. |
| [131] | Menelaou A (2013) Genotype calling and phasing using next- generation sequencing reads and a haplotype scaffold. Bioinformatics, 29, 84-91. |
| [132] | Meng C, Kubatko LS (2009) Detecting hybrid speciation in the presence of incomplete lineage sorting using gene tree incongruence: a model. Theoretical Population Biology, 75, 35-45. |
| [133] | Menozzi P, Piazza A, Cavallisforza L (1978) Synthetic maps of human gene frequencies in Europeans. Science, 201, 786-792. |
| [134] | Meyer M, Kircher M, Gansauge MT, Li H, Racimo F, Mallick S, Schraiber JG, Jay F, Prüfer K, de Filippo C (2012) A high-coverage genome sequence from an archaic Denisovan individual. Science, 338, 222-226. |
| [135] | Meyer M, Arsuaga JL, Filippo CD, Nagel S, Aximupetri A, Nickel B, Martínez I, Gracia A, Castro JMBD, Carbonell E (2016) Nuclear DNA sequences from the Middle Pleistocene Sima de los Huesos hominins. Nature, 531, 504-507. |
| [136] | Meyer M, Stenzel U, Hofreiter M (2008) Parallel tagged sequencing on the 454 platform. Nature Protocol, 3, 267-278. |
| [137] | Misof B, Misof K (2009) A Monte Carlo approach successfully identifies randomness in multiple sequence alignments: a more objective means of data exclusion. Systematic Biology, 58, 21-34. |
| [138] | Moorjani P, Patterson N, Hirschhorn JN, Keinan A, Hao L, Atzmon G, Burns E, Ostrer H, Price AL, Reich D (2011) The history of African gene flow into southern Europeans, Levantines, and Jews. PLoS Genetics, 7, e1001373. |
| [139] | Moreno-Estrada A, Gravel S, Zakharia F, Mccauley JL, Byrnes JK, Gignoux CR, Ortiz-Tello PA, Martínez RJ, Hedges DJ, Morris RW (2013) Reconstructing the population genetic history of the Caribbean. PLoS Genetics, 9, 569-573. |
| [140] | Morgan JAT, Harry AV, Welch DJ, Street R, White J, Geraghty PT, Macbeth WG, Tobin A, Simpfendorfer CA, Ovenden JR (2012) Detection of interspecies hybridisation in Chondrichthyes: hybrids and hybrid offspring between Australian (Carcharhinus tilstoni) and common (C. limbatus) blacktip shark found in an Australian fishery. Conservation Genetics, 13, 455-463. |
| [141] | Morin PA, Archer FI, Foote AD, Vilstrup J, Allen EE, Wade P, Durban J, Parsons K, Pitman R, Li L (2010) Complete mitochondrial genome phylogeographic analysis of killer whales (Orcinus orca) indicates multiple species. Genome Research, 20, 908-916. |
| [142] | Morrison DA (2011) Estimating species trees: practical and theoretical aspects. Systematic Biology, 60, 562-564. |
| [143] | Nabholz B, Künstner A, Wang R, Jarvis ED, Ellegren H (2011) Dynamic evolution of base composition: causes and consequences in avian phylogenomics. Molecular Biology and Evolution, 28, 2197. |
| [144] | Nakhleh L (2013) Computational approaches to species phylogeny inference and gene tree reconciliation. Trends in Ecology and Evolution, 28, 719-728. |
| [145] | Nelson RR (1963) Interspecific hybridization in the fungi. Annual Reviews in Microbiology, 17, 31-48. |
| [146] | Nielsen R, Paul JS, Albrechtsen A, Song YS (2011) Genotype and SNP calling from next-generation sequencing data. Nature Reviews Genetics, 12, 443-451. |
| [147] | Nosil P, Schluter D (2011) The genes underlying the process of speciation. Trends in Ecology and Evolution, 26, 160-167. |
| [148] | Novikova PY, Hohmann N, Nizhynska V, Tsuchimatsu T, Ali J, Muir G, Guggisberg A, Paape T, Schmid K, Fedorenko OM, Holm S, Sall T, Schlotterer C, Marhold K, Widmer A, Sese J, Shimizu KK, Weigel D, Kramer U, Koch MA, Nordborg M (2016) Sequencing of the genus Arabidopsis identifies a complex history of nonbifurcating speciation and abundant trans-specific polymorphism. Nature Genetics, 48, 1077-1082. |
| [149] | Omberg L, Salit J, Hackett N, Fuller J, Matthew R, Chouchane L, Rodriguez-Flores JL, Bustamante C, Crystal RG, Mezey JG (2012) Inferring genome-wide patterns of admixture in Qataris using fifty-five ancestral populations. BMC Genetics, 13, 49-59. |
| [150] | O’Neill EM, Schwartz R, Bullock CT, Williams JS, Shaffer HB, Aguilar-Miguel X, Parra-Olea G, Weisrock DW (2013) Parallel tagged amplicon sequencing reveals major lineages and phylogenetic structure in the North American tiger salamander (Ambystoma tigrinum) species complex. Molecular Ecology, 22, 111-129. |
| [151] | Ozsolak F (2011) RNA sequencing: advances, challenges and opportunities. Nature Reviews Genetics, 12, 87-98. |
| [152] | Padhukasahasram B (2014) Inferring ancestry from population genomic data and its applications. Frontiers in Genetics, 5, doi: 10.3389/fgene.2014.00204. |
| [153] | Pagel M, Meade A (2004) A phylogenetic mixture model for detecting pattern-heterogeneity in gene sequence or character-state data. Systematic Biology, 53, 571-581. |
| [154] | Pagel M, Meade A (2008) Modelling heterotachy in phylogenetic inference by reversible-jump Markov chain Monte Carlo. Philosophical Transactions of the Royal Society of London, 363, 3955-3964. |
| [155] | Parks M (2009) Increasing phylogenetic resolution at low taxonomic levels using massively parallel sequencing of chloroplast genomes. BMC Biology, 7, doi: 10.1186/1741- 7007-7-84. |
| [156] | Pasaniuc B, Sankararaman S, Kimmel G, Halperin E (2009) Inference of locus-specific ancestry in closely related populations. Bioinformatics, 25, 213-221. |
| [157] | Patterson N, Moorjani P, Luo YT, Mallick S, Rohland N, Zhan YP, Genschoreck T, Webster T, Reich D (2012) Ancient admixture in human history. Genetics, 192, 1065-1093. |
| [158] | Pavy N, Gagnon F, Deschênes A, Boyle B, Beaulieu J, Bousquet J (2016) Development of highly reliable in silico SNP resource and genotyping assay from exome capture and sequencing: an example from black spruce (Picea mariana). Molecular Ecology Resources, 16, 588-598. |
| [159] | Payseur BA, Rieseberg LH (2016) A genomic perspective on hybridization and speciation. Molecular Ecology, 25, 2337-2360. |
| [160] | Pease JB, Haak DC, Hahn MW, Moyle LC (2016) Phylogenomics reveals three sources of adaptive variation during a rapid radiation. PLoS Biology, 14, e1002379. |
| [161] | Peterson BK, Weber JN, Kay EH, Fisher HS, Hoekstra HE (2012) Double digest RADseq: an inexpensive method for de novo SNP discovery and genotyping in model and non-model species. PLoS ONE, 7, e37135. |
| [162] | Pérez-Losada M, Arenas M, Galán JC, Palero F, González-Candelas F (2015) Recombination in viruses: mechanisms, methods of study, and evolutionary consequences. Infection, Genetics and Evolution, 30, 296-307. |
| [163] | Philippe H, Delsuc F, Brinkmann H, Lartillot N (2005) Phylogenomics. Annual Review of Ecology, Evolution, and Systematics, 36, 541-562. |
| [164] | Pickrell JK, Pritchard JK (2012) Inference of population splits and mixtures from genome-wide allele frequency data. PLoS Genetics, 8, e1002967. |
| [165] | Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D (2006) Principal components analysis corrects for stratification in genome-wide association studies. Nature Genetics, 38, 904-909. |
| [166] | Price AL, Tandon A, Patterson N, Barnes KC, Rafaels N, Ruczinski I, Beaty TH, Mathias R, Reich D, Myers S (2009) Sensitive detection of chromosomal segments of distinct ancestry in admixed populations. PLoS Genetics, 5, e1000519. |
| [167] | Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics, 155, 945-959. |
| [168] | Prufer K, Racimo F, Patterson N, Jay F, Sankararaman S, Sawyer S, Heinze A, Renaud G, Sudmant PH, de Filippo C, Li H, Mallick S, Dannemann M, Fu QM, Kircher M, Kuhlwilm M, Lachmann M, Meyer M, Ongyerth M, Siebauer M, Theunert C, Tandon A, Moorjani P, Pickrell J, Mullikin JC, Vohr Samuel H, Green RE, Hellmann I, Johnson PLF, Blanche H, Cann H, Kitzman JO, Shendure J, Eichler EE, Lein ES, Bakken TE, Golovanova LV, Doronichev VB, Shunkov MV, Derevianko AP, Viola B, Slatkin M, Reich D, Kelso J, Paabo S (2014) The complete genome sequence of a Neanderthal from the Altai Mountains. Nature, 505, 43-49. |
| [169] | Pyron RA, Hsieh FW, Lemmon AR, Lemmon EM, Hendry CR (2016) Integrating phylogenomic and morphological data to assess candidate species-delimitation models in brown and red-bellied snakes (Storeria). Zoological Journal of the Linnean Society, 177, 937-949. |
| [170] | Raghavan M, DeGiorgio M, Albrechtsen A, Moltke I, Skoglund P, Korneliussen TS, Gronnow B, Appelt M, Gullov HC, Friesen TM, Fitzhugh W, Malmstrom H, Rasmussen S, Olsen J, Melchior L, Fuller BT, Fahrni SM, Stafford TJ, Grimes V, Renouf MA, Cybulski J, Lynnerup N, Lahr MM, Britton K, Knecht R, Arneborg J, Metspalu M, Cornejo OE, Malaspinas AS, Wang Y, Rasmussen M, Raghavan V, Hansen TV, Khusnutdinova E, Pierre T, Dneprovsky K, Andreasen C, Lange H, Hayes MG, Coltrain J, Spitsyn VA, Gotherstrom A, Orlando L, Kivisild T, Villems R, Crawford MH, Nielsen FC, Dissing J, Heinemeier J, Meldgaard M, Bustamante C, O’Rourke DH, Jakobsson M, Gilbert MT, Nielsen R, Willerslev E (2014a) The genetic prehistory of the new world Arctic. Science, 345, doi: 10.1126/science.1255832. |
| [171] | Raghavan M, Skoglund P, Graf KE, Metspalu M, Albrechtsen A, Moltke I, Rasmussen S, Stafford TW Jr, Orlando L, Metspalu E (2014) Upper Palaeolithic Siberian genome reveals dual ancestry of native Americans. Nature, 505, 87-91. |
| [172] | Rannala B, Yang Z (2008) Phylogenetic inference using whole genomes. Annual Review of Genomics & Human Genetics, 9, 217-231. |
| [173] | Rasmussen MD, Hubisz MJ, Gronau I, Siepel A (2014) Genome-wide inference of ancestral recombination graphs. PLoS Genetics, 10, e1004342. |
| [174] | Rasmussen MD, Kellis M (2012) Unified modeling of gene duplication, loss, and coalescence using a locus tree. Genome Research, 22, 755-765. |
| [175] | Reich D, Thangaraj K, Patterson N, Price AL, Singh L (2009) Reconstructing Indian population history. Nature, 461, 489-494. |
| [176] | Reich D, Green RE, Kircher M, Krause J, Patterson N, Durand EY, Viola B, Briggs AW, Stenzel U, Johnson PLF (2010) Genetic history of an archaic hominin group from Denisova Cave in Siberia. Nature, 468, 1053-1060. |
| [177] | Reneker J, Lyons E, Conant GC, Pires JC, Freeling M, Shyu CR, Korkin D (2012) Long identical multispecies elements in plant and animal genomes. Proceedings of the National Academy of Sciences, USA, 109, 1183-1191. |
| [178] | Rieseberg LH, Archer MA, Wayne RK (1999) Transgressive segregation, adaptation and speciation. Heredity, 83, 363-372. |
| [179] | Rieseberg LH, Widmer A, Arntz AM, Burke B (2003) The genetic architecture necessary for transgressive segregation is common in both natural and domesticated populations. Philosophical Transactions of the Royal Society of London Series B: Biological Sciences, 358, 1141-1147. |
| [180] | Robinson JD, Bunnefeld L, Hearn J, Stone GN, Hickerson MJ (2014) ABC inference of multi-population divergence with admixture from unphased population genomic data. Molecular Ecology, 23, 4458-4471. |
| [181] | Rodríguezezpeleta N, Brinkmann H, Roure B, Lartillot N, Lang BF, Philippe H (2007) Detecting and overcoming systematic errors in genome-scale phylogenies. Systematic Biology, 56, 389-399. |
| [182] | Rodriguez JM, Bercovici S, Elmore M, Batzoglou S (2013) Ancestry inference in complex admixtures via variable- length Markov chain linkage models. Journal of Computational Biology, 20, 199-211. |
| [183] | Rusk N (2009) Focus on next-generation sequencing data analysis. Nature Methods, 6, doi: 10.1038/nmeth.f.271. |
| [184] | Sankararaman S, Patterson N, Li H, Pääbo S, Reich D (2012) The date of interbreeding between Neandertals and modern humans. PLoS Genetics, 8, e1002947. |
| [185] | Sankararaman S, Mallick S, Dannemann M, Prufer K, Kelso J, Paabo S, Patterson N, Reich D (2014) The genomic landscape of Neanderthal ancestry in present-day humans. Nature, 507, 354-357. |
| [186] | Sankararaman S, Sridhar S, Kimmel G, Halperin E (2008) Estimating local ancestry in admixed populations. American Journal of Human Genetics, 82, 290-303. |
| [187] | Schmickl R, Liston A, Zeisek V, Oberlander K, Weitemier K, Straub SCK, Cronn RC, Dreyer LL, Suda J (2016) Phylogenetic marker development for target enrichment from transcriptome and genome skim data: the pipeline and its application in southern African Oxalis (Oxalidaceae). Molecular Ecology Resources, 16, 1124-1135. |
| [188] | Schwenk K, Brede N, Streit B (2008) Introduction: extent, processes and evolutionary impact of interspecific hybridization in animals. Philosophical Transactions of the Royal Society B: Biological Sciences, 363, 2805-2811. |
| [189] | Shafer ABA, Peart CR, Tusso S, Maayan I, Brelsford A, Wheat CW, Wolf JBW (2016) Bioinformatic processing of RAD- seq data dramatically impacts downstream population genetic inference. Methods in Ecology and Evolution, doi: 10.1111/2041-210X.12700. |
| [190] | Shen XX, Liang D, Feng YJ, Chen MY, Zhang P (2013) A versatile and highly efficient toolkit including 102 nuclear markers for vertebrate phylogenomics, tested by resolving the higher level relationships of the caudata. Molecular Biology and Evolution, 30, 2235-2248. |
| [191] | Siepel A (2009) Phylogenomics of primates and their ancestral populations. Genome Research, 19, 1929-1941. |
| [192] | Sims GE, Jun SR, Wu GA, Kim SH (2009) Alignment-free genome comparison with feature frequency profiles (FFP) and optimal resolutions. Proceedings of the National Academy of Sciences, USA, 106, 2677-2682. |
| [193] | Skotte L, Korneliussen TS, Albrechtsen A (2013) Estimating individual admixture proportions from next generation sequencing data. Genetics, 195, 693-702. |
| [194] | Smith J, Kronforst MR (2013) Do Heliconius butterfly species exchange mimicry alleles? Biology Letters, 9, 20130503. |
| [195] | Sneath PHA (1975) Cladistic representation of reticulate evolution. Systemtic Zoology, 24, 360-368. |
| [196] | Solis-Lemus C, Ane C (2016) Inferring phylogenetic networks with maximum pseudolikelihood under incomplete lineage sorting. PLoS Genetics, 12, e1005896. |
| [197] | Soltis PS, Soltis DE (2009) The role of hybridization in plant speciation. Annual Review of Plant Biology, 60, 561-588. |
| [198] | Song BH, Wang XQ, Wang XR, Ding KY, Hong DY (2003) Cytoplasmic composition in Pinus densata and population establishment of the diploid hybrid pine. Molecular Ecology, 12, 2995-3001. |
| [199] | Song BH, Wang XQ, Wang XR, Sun LJ, Hong DY, Peng PH (2002) Maternal lineages of Pinus densata, a diploid, hybrid. Molecular Ecology, 11, 1057-1063. |
| [200] | Song YS, Hein J (2005) Constructing minimal ancestral recombination graphs. Journal of Computational Biology, 12, 147-169. |
| [201] | Stenz NWM, Larget B, Baum DA, Ané C (2015) Exploring tree-like and non-tree-like patterns using genome sequences: an example using the inbreeding plant species Arabidopsis thaliana (L.) Heynh. Systematic Biology, 64, 809-823. |
| [202] | Strimmer K, Moulton V (2000) Likelihood analysis of phylogenetic networks using directed graphical models. Molecular Biology and Evolution, 17, 875-881. |
| [203] | Su S, Wong G, Shi WF, Liu J, Lai ACK, Zhou JY, Liu WJ, Bi YH, Gao GF (2016) Epidemiology, genetic recombination, and pathogenesis of coronaviruses. Trends in Microbiology, 24, 490-502. |
| [204] | Sullivan J, Joyce P (2005) Model selection in phylogenetics. Annual Review of Ecology, Evolution, and Systematics, 36, 445-466. |
| [205] | Sundquist A, Fratkin E, Do CB, Batzoglou S (2008) Effect of genetic divergence in identifying ancestral origin using HAPAA. Genome Research, 18, 676-682. |
| [206] | Sunnåker M, Busetto AG, Numminen E, Corander J, Foll M, Dessimoz C (2013) Approximate Bayesian computation. PLoS Computational Biology, 9, e1002803. |
| [207] | Susko E, Spencer M, Roger AJ (2005) Biases in phylogenetic estimation can be caused by random sequence segments. Journal of Molecular Evolution, 61, 351-359. |
| [208] | Swofford DL, Olsen GJ, Waddell PJ, Hillis DM (1996) Phylogenetic inference. In: Molecular Systematics (eds Hiillis DM, Moritz D, Mable BK), pp. 407-514. Sinauer Associates, Sunderland, Massachusetts. |
| [209] | Szymura JM, Barton NH (1986) Genetic analysis of a hybrid zone between the fire-bellied toads, Bombina bombina and B. variegata, near Cracow in southern Poland. Evolution, 40, 1141-1159. |
| [210] | Tang H, Choudhry S, Mei R, Morgan M, Rodriguez-Cintron W, Burchard EG, Risch NJ (2007) Recent genetic selection in the ancestral admixture of Puerto Ricans. American Journal of Human Genetics, 81, 626-633. |
| [211] | Tang H, Coram M, Wang P, Zhu X, Risch N (2006) Reconstructing genetic ancestry blocks in admixed individuals. American Journal of Human Genetics, 79, 1-12. |
| [212] | Than C, Ruths D, Nakhleh L (2008) PhyloNet: a software package for analyzing and reconstructing reticulate evolutionary relationships. BMC Bioinformatics, 9, doi: 10.1186/ 1471-2105-9-322. |
| [213] | The Genomes Project Consortium, Altshuler DL, Durbin RM, Abecasis GR, Bentley DR, Chakravarti A, Clark AG, Collins FS, de La Vega FM (2010) A map of human genome variation from population-scale sequencing. Nature, 467, 1061-1073. |
| [214] | Torkamaneh D, Laroche J, Bastien M, Abed A, Belzile F (2017) Fast-GBS: a new pipeline for the efficient and highly accurate calling of SNPs from genotyping-by-sequencing data. BMC Bioinformatics, 18, doi: 10.1186/s12859-016- 1431-9. |
| [215] | Torkamaneh D, Laroche J, Belzile F (2016) Genome-wide SNP calling from genotyping by sequencing (GBS) data: a comparison of seven pipelines and two sequencing technologies. PLoS ONE, 11, e0161333. |
| [216] | Townsend JP (2007) Profiling phylogenetic informativeness. Systematic Biology, 56, 222-231. |
| [217] | Vallejo-Marin M, Hiscock SJ (2016) Hybridization and hybrid speciation under global change. New Phytologist, 211, 1170-1187. |
| [218] | van Tassell CP, Smith TP, Matukumalli LK, Taylor JF, Schnabel RD, Lawley CT, Haudenschild CD, Moore SS, Warren WC, Sonstegard TS (2008) SNP discovery and allele frequency estimation by deep sequencing of reduced representation libraries. Nature Methods, 5, 247-252. |
| [219] | Wang BS, Mao JF, Gao J, Zhao W, Wang XR (2011) Colonization of the Tibetan Plateau by the homoploid hybrid pine Pinus densata. Molecular Ecology, 20, 3796-3811. |
| [220] | Wang S, Meyer E, Mckay JK, Matz MV (2012) 2b-RAD: a simple and flexible method for genome-wide genotyping. Nature Methods, 9, 808-810. |
| [221] | Wang XR, Szmidt AE (1994) Hybridization and chloroplast DNA variation in a Pinus species complex from Asia. Evolution, 48, 1020-1031. |
| [222] | Wang XR, Szmidt AE, Savolainen O (2001) Genetic composition and diploid hybrid speciation of a high mountain pine, Pinus densata, native to the Tibetan Plateau. Genetics, 159, 337-346. |
| [223] | Wang XR, Szmidt AE, Lewandowski A, Wang ZR (1990) Evolutionary analysis of Pinus densata Masters, a putative tertiary hybrid 1, allozyme variation. Theoretical and Applied Genetics, 80, 635-640. |
| [224] | Wang Z, Gerstein M, Snyder M (2009) RNA-Seq: a revolutionary tool for transcriptomics. Nature Reviews Genetics, 10, 57-63. |
| [225] | Wegmann D, Leuenberger C, Neuenschwander S, Excoffier L (2010) ABCtoolbox: a versatile toolkit for approximate Bayesian computations. BMC Bioinformatics, 11, doi:10.1186/ 1471-2105-11-116. |
| [226] | Weigel D, Mott R (2009) The 1001 Genomes Project for Arabidopsis thaliana. Genome Biology, 10, 107. |
| [227] | Worobey M, Holmes EC (1999) Evolutionary aspects of recombination in RNA viruses. Journal of General Virology, 80, 2535-2543. |
| [228] | Wu CL (1956) The taxonomic revision and phytogeographical study of Chinese pines. Acta Phytotaxonomica Sinica, 5, 131-163. (in Chinese with English abstract) |
| [吴中伦 (1956) 中国松属的分类与分布. 植物分类学报, 5, 131-163.] | |
| [229] | Wu CI, Ting CT (2004) Genes and speciation. Nature Reviews Genetics, 5, 114-122. |
| [230] | Wu Y (2012) Coalescent-based species tree inference from gene tree topologies under incomplete lineage sorting by maximum likelihood. Evolution, 66, 763-775. |
| [231] | Wu ZY, Raven PH, Hong DY (2014) Flora of China, Vols. 1-25. Science Press, Beijing & Missouri Botanical Garden Press, St. Louis. |
| [232] | Xiang YZ, Huang CH, Hu Y, Wen J, Li SS, Yi TS, Chen HY, Xiang J, Ma H (2017) Well-resolved rosaceae nuclear phylogeny facilitates geological time and genome duplication analyses and ancestral fruit character reconstruction. Molecular Biology and Evolution, 34, 262-281. |
| [233] | Xing FQ, Mao JF, Meng JX, Dai JF, Zhao W, Liu H, Xing Z, Zhang H, Wang XR, Li Y (2014) Needle morphological evidence of the homoploid hybrid origin of Pinus densata based on analysis of artificial hybrids and the putative parents, Pinus tabuliformis and Pinus yunnanensis. Ecology & Evolution, 4, 1890-1902. |
| [234] | Yang JJ, Li J, Buu A, Williams LK (2013) Efficient inference of local ancestry. Bioinformatics, 29, 2750-2756. |
| [235] | Yang WY, Novembre J, Eskin E, Halperin E (2012) A model-based approach for analysis of spatial structure in genetic data. Nature Genetics, 44, 725-731. |
| [236] | Yang ZH (1996) Maximum-likelihood models for combined analyses of multiple sequence data. Journal of Molecular Evolution, 42, 587-596. |
| [237] | Yu Y, Degnan JH, Nakhleh L (2012) The probability of a gene tree topology within a phylogenetic network with applications to hybridization detection. PLoS Genetics, 8, e1002660. |
| [238] | Yu Y, Dong J, Liu KJ, Nakhleh L (2014) Maximum likelihood inference of reticulate evolutionary histories. Proceedings of the National Academy of Sciences, USA, 111, 16448-16453. |
| [239] | Yu Y, Ristic N, Nakhleh L (2013) Fast algorithms and heuristics for phylogenomics under ILS and hybridization. BMC Bioinformatics, 14, 1-10. |
| [240] | Yu Y, Than C, Degnan JH, Nakhleh L (2011) Coalescent histories on phylogenetic networks and detection of hybridization despite incomplete lineage sorting. Systematic Biology, 60, 138-149. |
| [241] | Yu Y, Nakhleh L (2015) A maximum pseudo-likelihood approach for phylogenetic networks. BMC Genomics, 16, 1-10. |
| [242] | Zhang LS, Dai JF, Gao Q, Liu H, Zhang H, Zhao W, Mao JF, Li Y (2012) Seedling adaptation of hybrid pine Pinus densata and its parental species in the high elevation habitat. Journal of Beijing Forestry University, 34(5), 15-24. (in Chinese with English abstract) |
| [张立沙, 代剑峰, 高琼, 刘灏, 张华, 赵伟, 毛建丰, 李悦 (2012) 高山松与亲本种多种群在高海拔生境下的苗期适应性研究. 北京林业大学学报, 34(5), 15-24.] | |
| [243] | Zeng LP, Zhang Q, Sun RR, Kong HZ, Zhang N, Ma H (2014) Resolution of deep angiosperm phylogeny using conserved nuclear genes and estimates of early divergence times. Nature Communications, 5, doi: 10.1038/ncomms5956. |
| [244] | Zhao W, Meng JX, Wang BS, Zhang LS, Xu YL, Zeng QY, Li Y, Mao JF, Wang XR (2014) Weak crossability barrier but strong juvenile selection supports ecological speciation of the hybrid pine Pinus densata on the Tibetan Plateau. Evolution, 68, 3120-3133. |
| [245] | Zheng XH, Lu F, Wang ZY, Zhong F, Hoover J, Mural R (2005) Using shared genomic synteny and shared protein functions to enhance the identification of orthologous gene pairs. Bioinformatics, 21, 703-710. |
| [1] | Lijie Zhou, Minhui Huang, Huaijiang He, Yanxia Cheng, Chunyu Zhang, Xiuhai Zhao. Disentangling the patterns, components, and influencing factors of forest β diversity in the Xiaoxing’an Mountains, Northeast China [J]. Biodiv Sci, 2026, 34(4): 25443-. |
| [2] | Yunyue Peng, Kui Peng, Xinran Liu, Tianyi Sun, Xiaoquan Zhang. Corporate engagement pathways, market development barriers and recommendations for biodiversity credits [J]. Biodiv Sci, 2026, 34(4): 26031-. |
| [3] | Boyao Li, Tiancheng Sheng, Xiaoyun Xing. Impact of biodiversity risk on corporate financial performance: Evidence from listed companies in China [J]. Biodiv Sci, 2026, 34(4): 25330-. |
| [4] | Hao Liu, Yuxiao Zhang, Bing Liu, Feifei Li, Hongzheng Ma, Haining Qin, Dezhu Li, Wenli Chen. Diversity and checklist of grasses (Poaceae) in China [J]. Biodiv Sci, 2026, 34(4): 25438-. |
| [5] | Ziyi Kong, Degang Wang, Jiantao Wang, Zhiyong Pei, Jing Sun, Changchun Zhang, Junguo Zhang. Wildlife pose estimation based on the SCD-HRNet model and its application in biodiversity monitoring: A case study of the Saihanwula Region, Inner Mongolia [J]. Biodiv Sci, 2026, 34(4): 25287-. |
| [6] | Haoyou Zhu, Youbing Zhou, Yi Luo, Zhaomin Zhou. Changes in breeding bird community in an urban area of Nanchong over two decades [J]. Biodiv Sci, 2026, 34(3): 24560-. |
| [7] | Pengfei Fan. Research advances and conservation of gibbons in China [J]. Biodiv Sci, 2026, 34(3): 25456-. |
| [8] | Shuyu Hou, Yingying Liu, Rui Yang. Progress of international OECMs practices under the Kunming-Montreal Global Biodiversity Framework and pathways for localization in China [J]. Biodiv Sci, 2026, 34(3): 25264-. |
| [9] | Xiaofan Cheng, Qingyuan Li, Yuanhui Li, Mingxiang Zhang. The dilemmas and solutions for invasive alien species governance policy systems [J]. Biodiv Sci, 2026, 34(2): 25332-. |
| [10] | Lulu Chen, Haoting Tang, Hong Leng, Qing Yuan, Xinyue Yang. Impacts of urban block built environments on biodiversity—A review and outlook [J]. Biodiv Sci, 2026, 34(2): 25286-. |
| [11] | Wenqi Gao, Jingrong Xiang, Yao Zhao, Lingshuang Fan, Yuan Gu, Weihan Shao, Gaojun Li, Guangjun Zhao, Mingbin Chen, Xingwei Cai, Kai Chen. Stream fish community characteristics and their response to land use in Limushan and Jianfengling of Hainan Tropical Rainforest National Park [J]. Biodiv Sci, 2026, 34(2): 25374-. |
| [12] | Xiaoqiang Lu, Dan Rui, Jiangfeng Zhang, Bingxin Yin, Yulu Wang, Yuting Cen, Yichen Cui, Wanxia Yang. Impacts of nitrogen inputs-driven key ecological processes on biodiversity and their management implications [J]. Biodiv Sci, 2026, 34(2): 25368-. |
| [13] | Tinghong Tan, Fan Gao, Yu Yang, Qunying Xiao, Chunfang Wu, Na Qiu, Ningning Zhao, Min Zhou, Gongping Kang, Zhihong Lu, Jianqiang Gao, Hong Yang, Chuandong Yang, Chunying Deng. Macrofungal species cataloging in karst areas of southwestern China [J]. Biodiv Sci, 2026, 34(2): 25281-. |
| [14] | Xuri Zhang, Biao Luo, Tong Zhao, Dan Huang, Weiming Ai. Fish diversity of Zhejiang Province: Inventory, distribution and conservation [J]. Biodiv Sci, 2026, 34(2): 25225-. |
| [15] | Haiou Liu, Zhiming Hao, Leshan Du, Wenhui Liu, Ziyuan Li, Lei Liu. Progress, challenges, and insights on the operation of the Global Biodiversity Framework Fund [J]. Biodiv Sci, 2026, 34(2): 25463-. |
| Viewed | ||||||
|
Full text |
|
|||||
|
Abstract |
|
|||||
Copyright © 2026 Biodiversity Science
Editorial Office of Biodiversity Science, 20 Nanxincun, Xiangshan, Beijing 100093, China
Tel: 010-62836137, 62836665 E-mail: biodiversity@ibcas.ac.cn