
Biodiv Sci ›› 2026, Vol. 34 ›› Issue (1): 25263. DOI: 10.17520/biods.2025263 cstr: 32101.14.biods.2025263
• Special Feature: Methods for Ecological Data Analysis • Previous Articles Next Articles
Yuan Jiang1, Beixi Huang1, Xueyuan Jia1, Si Liang1, Yutong Xie1, Ping Fan1,*( )(
)( ), Gang Song2()
), Gang Song2()
Received:2025-07-06
Accepted:2025-09-30
Online:2026-01-20
Published:2026-01-22
Contact:
Ping Fan
Supported by:Yuan Jiang, Beixi Huang, Xueyuan Jia, Si Liang, Yutong Xie, Ping Fan, Gang Song. An approach for estimating haplotype richness from DNA sequences with unequal lengths[J]. Biodiv Sci, 2026, 34(1): 25263.
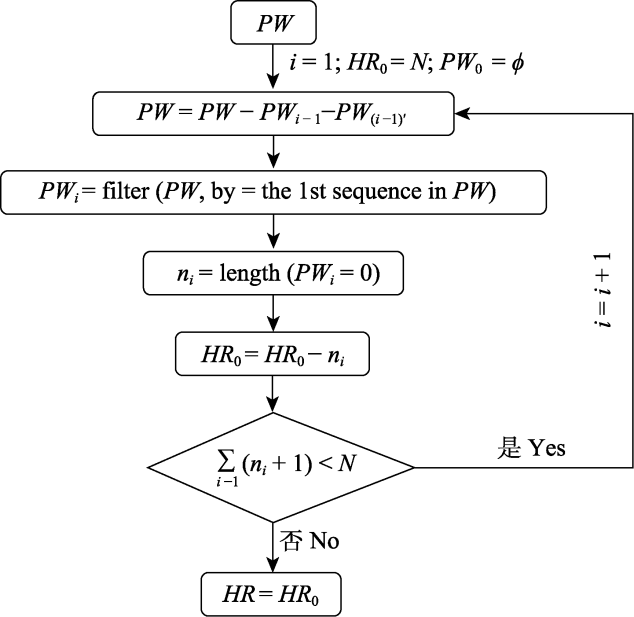
Fig. 1 The computational framework of haplotype richness
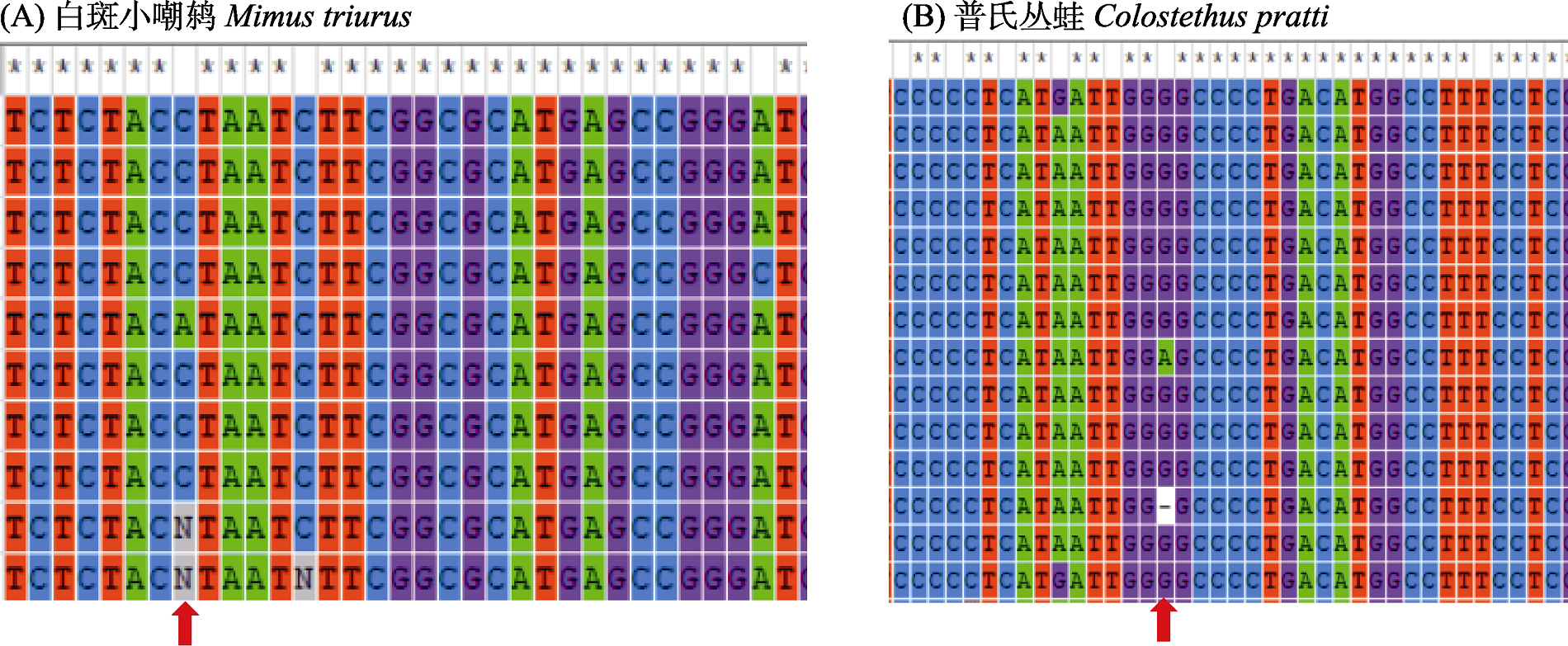
Fig. 2 An example of species data for sequences of equal length with missing bases (partial range). Red arrows indicate the nucleotide segregation sites where missing bases are found.
| 伪R2 Pseudo R2 | 一次项系数 β1 | 二次项系数 β2 | |
|---|---|---|---|
| 鸟类 Birds | 0.280 | 1.458e-02*** | -5.116e-04*** |
| 哺乳动物 Mammals | 0.361 | 1.788e-02*** | -5.506e-04*** |
| 两栖动物 Amphibians | 0.518 | -8.046e-02*** | 4.659e-04*** |
Table 1 Beta regression models for latitudinal gradients in haplotype richness of birds, mammals, and amphibians
| 伪R2 Pseudo R2 | 一次项系数 β1 | 二次项系数 β2 | |
|---|---|---|---|
| 鸟类 Birds | 0.280 | 1.458e-02*** | -5.116e-04*** |
| 哺乳动物 Mammals | 0.361 | 1.788e-02*** | -5.506e-04*** |
| 两栖动物 Amphibians | 0.518 | -8.046e-02*** | 4.659e-04*** |
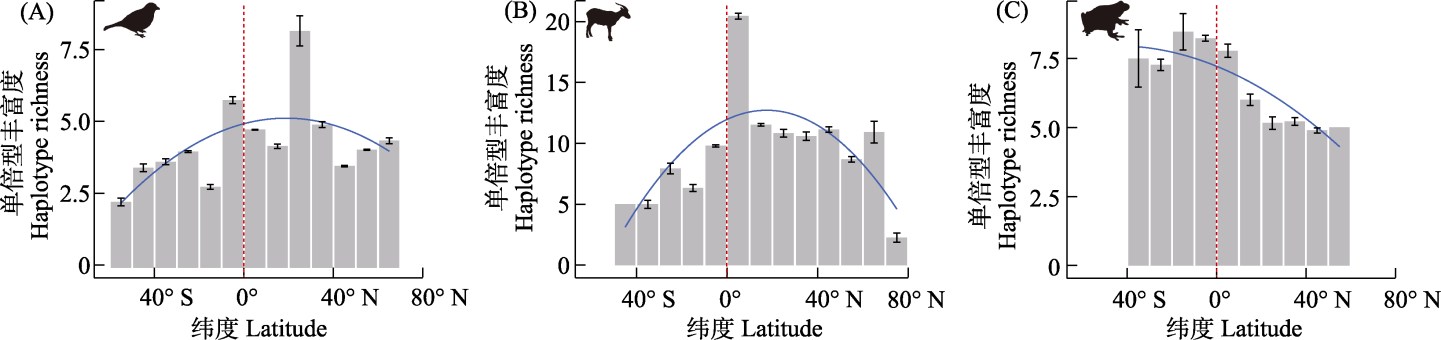
Fig. 3 The latitudinal gradients in haplotype richness of birds (A), mammals (B), and amphibians (C). The regression lines (blue lines) represent the predictions of the beta regression model. The red dashed lines show the equator.
| [1] |
Chang YB, Song G, Zhang DZ, Jia CX, Fan P, Hao Y, Ji YZ, Lei FM (2022) Distribution pattern and driving factors of genetic diversity of passerine birds in the mountains of Southwest China. Avian Research, 13, 100043.
DOI URL |
| [2] |
Edgar RC (2022) Muscle5: High-accuracy alignment ensembles enable unbiased assessments of sequence homology and phylogeny. Nature Communications, 13, 6968.
DOI PMID |
| [3] |
Ellegren H, Galtier N (2016) Determinants of genetic diversity. Nature Reviews Genetics, 17, 422-433.
DOI PMID |
| [4] |
Fan P, Fjeldså J, Liu X, Dong YF, Chang YB, Qu YH, Song G, Lei FM (2021) An approach for estimating haplotype diversity from sequences with unequal lengths. Methods in Ecology and Evolution, 12, 1658-1667.
DOI URL |
| [5] |
Fan P, Song G, Qiao HJ, Zhang DZ, Ji YZ, Qu YH, Fjeldså J, Lei FM (2025) Revaluation of the genetic diversity-area relationship by integrating nucleotide and haplotype diversity. Current Zoology, 71, 645-651.
DOI URL |
| [6] |
Ferrari S, Cribari-Neto F (2004) Beta regression for modelling rates and proportions. Journal of Applied Statistics, 31, 799-815.
DOI URL |
| [7] |
Fordham DA, Saltré F, Haythorne S, Wigley TML, Otto- Bliesner BL, Chan KC, Brook BW (2017) PaleoView: A tool for generating continuous climate projections spanning the last 21000 years at regional and global scales. Ecography, 40, 1348-1358.
DOI URL |
| [8] |
Goodall-Copestake WP, Tarling GA, Murphy EJ (2012) On the comparison of population-level estimates of haplotype and nucleotide diversity: A case study using the gene cox1 in animals. Heredity, 109, 50-56.
DOI PMID |
| [9] |
Gratton P, Marta S, Bocksberger G, Winter M, Keil P, Trucchi E, Kühl H (2017) Which latitudinal gradients for genetic diversity? Trends in Ecology & Evolution, 32, 724-726.
DOI URL |
| [10] | Hijmans RJ, Williams E, Vennes C (2019) geosphere: Spherical Trigonometry. R Package Version 1.5-10, https://CRAN.R-project.org/package=geosphere. (accessed on 2021-05-21) |
| [11] |
Hong B, Rabassa J, Uchida M, Hong YT, Peng HJ, Ding HW, Guo Q, Yao H (2019) Response and feedback of the Indian summer monsoon and the Southern Westerly Winds to a temperature contrast between the hemispheres during the last glacial-interglacial transitional period. Earth-Science Reviews, 197, 102917.
DOI URL |
| [12] |
Leitwein M, Duranton M, Rougemont Q, Gagnaire PA, Bernatchez L (2020) Using haplotype information for conservation genomics. Trends in Ecology & Evolution, 35, 245-258.
DOI URL |
| [13] |
Librado P, Rozas J (2009) DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics, 25, 1451-1452.
DOI PMID |
| [14] | Mastretta-Yanes A, da Silva JM, Grueber CE, Castillo-Reina L, Köppä V, Forester BR, Funk WC, Heuertz M, Ishihama F, Jordan R, Mergeay J, Paz-Vinas I, Rincon-Parra VJ, Rodriguez-Morales MA, Arredondo-Amezcua L, Brahy G, DeSaix M, Durkee L, Hamilton A, Hunter ME, Koontz A, Lang I, Latorre-Cárdenas MC, Latty T, Llanes-Quevedo A, MacDonald AJ, Mahoney M, Miller C, Ornelas JF, Ramírez-Barahona S, Robertson E, Russo IM, Santiago MA, Shaw RE, Shea GM, Sjögren-Gulve P, Spence ES, Stack T, Suárez S, Takenaka A, Thurfjell H, Turbek S, van der Merwe M, Visser F, Wegier A, Wood G, Zarza E, Laikre L, Hoban S (2024) Multinational evaluation of genetic diversity indicators for the Kunming-Montreal Global Biodiversity Framework. Ecology Letters, 27, e14461. |
| [15] |
Miraldo A, Li S, Borregaard MK, Flórez-Rodríguez A, Gopalakrishnan S, Rizvanovic M, Wang ZH, Rahbek C, Marske KA, Nogués-Bravo D (2016) An Anthropocene map of genetic diversity. Science, 353, 1532-1535.
PMID |
| [16] | Nei M, Li WH (1979) Mathematical model for studying genetic variation in terms of restriction endonucleases. Proceedings of the National Academy of Sciences, USA, 76, 5269-5273. |
| [17] |
Nei M, Roychoudhury AK (1974) Sampling variances of heterozygosity and genetic distance. Genetics, 76, 379-390.
DOI PMID |
| [18] |
Nei M, Tajima F (1981) DNA polymorphism detectable by restriction endonucleases. Genetics, 97, 145-163.
DOI PMID |
| [19] |
Romiguier J, Gayral P, Ballenghien M, Bernard A, Cahais V, Chenuil A, Chiari Y, Dernat R, Duret L, Faivre N, Loire E, Lourenco JM, Nabholz B, Roux C, Tsagkogeorga G, Weber AAT, Weinert LA, Belkhir K, Bierne N, Glémin S, Galtier N (2014) Comparative population genomics in animals uncovers the determinants of genetic diversity. Nature, 515, 261-263.
DOI |
| [1] | Yang Qin, Aishan Wumaierjiang, Wentao Wei, Shamushake Diliniga. The impact of biodiversity information disclosure on supply chain resilience of A-share listed companies [J]. Biodiv Sci, 2026, 34(1): 25342-. |
| [2] | Guangtai Fan, Yining Chen, Jibai He, Hailong Dou, Jiyuan Chen, Haitao Yang, Qiu Shen, Hongcan Guan. Investigating the adaptation of Eurasian otters (Lutra lutra) to human disturbance and the coexistence with sympatric species using camera trapping: A case study from Jintang Island, Zhejiang Province, China [J]. Biodiv Sci, 2025, 33(9): 25148-. |
| [3] | Defu Chen, Mingrong Liang, Yijuan Xu. Overview of ant sampling methods in vertical spatial stratification [J]. Biodiv Sci, 2025, 33(8): 25078-. |
| [4] | Ruxiao Wang, Boyang Shi, Da Pan, Hongying Sun. Biodiversity patterns and conservation gaps of the endemic freshwater crab genus Sinopotamon in China [J]. Biodiv Sci, 2025, 33(8): 25123-. |
| [5] | Zhicheng Zhou, Tianling Cao, Ruyao Liu, Qiqi Ding, Ke Ma, Liping Yang, Chuanjiang Zhou, Guoxing Nie, Yongtao Tang. Genetic diversity and structure of Pseudorasbora parva population in the Yellow River basin based on the mitochondrial COI gene [J]. Biodiv Sci, 2025, 33(8): 24501-. |
| [6] | Aiying Wang, Wanjin Liao. Coevolutionary processes: Methods and advances in cophylogenetic analysis [J]. Biodiv Sci, 2025, 33(8): 25112-. |
| [7] | Qilong Yu, Minhui Hao, Huaijiang He, Chunyu Zhang, Xiuhai Zhao. Relationships of biodiversity and productivity change with forest succession in Changbai Mountains: Insights from species, traits, and phylogeny [J]. Biodiv Sci, 2025, 33(8): 25060-. |
| [8] | Ping Fan, Huan Wang, Zhixin Wen, Gang Song, Fuming Lei. Impact of climatic factors on the genetic diversity-species area relationship of birds [J]. Biodiv Sci, 2025, 33(8): 25072-. |
| [9] | Ruixiang Xue, Xuerong Ma, Jiongwen Wu, Aijun Liu, Xiquan Zhang, Congliang Ji, Yingshan Yin, Weijian Zhu, Qingbin Luo. Genetic diversity and genetic structure of Zhongshan partridge duck populations [J]. Biodiv Sci, 2025, 33(8): 24592-. |
| [10] | Ping Fan, Zhixin Wen, Gang Song. The effect of climatic factors and anthropogenic activities on different genetic diversity indicators of amphibians and mammals [J]. Biodiv Sci, 2025, 33(8): 25022-. |
| [11] | Hong Deyuan. A brief discussion on methodology in taxonomy [J]. Biodiv Sci, 2025, 33(2): 24541-. |
| [12] | Zhang Shuxin, Jia Zixuan, Fang Tao, Liu Yifan, Zhao Wei, Wang Rong, Chang Haichao, Luo Fangli, Zhu Yaojun, Yu Feihai. Methods to evaluate plant tolerance to environmental stresses [J]. Biodiv Sci, 2025, 33(2): 24168-. |
| [13] | Jinshan Wu, Changle Yang, Yufeng Ma, Yaxuan Li, Wenjia Gao, Ye Kusili, Lianghong Ye, Yujiao Yang, Mengqi Xu, Tingqiong Liao, Linqiang Zhong, Wenjuan Shan. Genetic diversity and genetic structure of red deer in the Ebinur Lake Wetland National Nature Reserve [J]. Biodiv Sci, 2025, 33(12): 25233-. |
| [14] | Jiachen Wang, Tangjun Xu, Wei Xu, Gaoji Zhang, Yijin You, Honghua Ruan, Hongyi Liu. Impact of urban landscape pattern on the genetic structure of Thereuopoda clunifera population in Nanjing, China [J]. Biodiv Sci, 2025, 33(1): 24251-. |
| [15] | Jiqi Gu, Jianping Chen, Jiangshan Lai. Application of large language models in biodiversity research [J]. Biodiv Sci, 2024, 32(9): 24258-. |
| Viewed | ||||||
|
Full text |
|
|||||
|
Abstract |
|
|||||
Copyright © 2026 Biodiversity Science
Editorial Office of Biodiversity Science, 20 Nanxincun, Xiangshan, Beijing 100093, China
Tel: 010-62836137, 62836665 E-mail: biodiversity@ibcas.ac.cn