|
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
联合物种分布模型与生物群落层次建模框架: 生态学理论、方法及应用
生物多样性
2026, 34 (1):
25364-.
DOI: 10.17520/biods.2025364
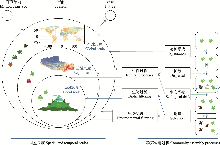
理解环境过滤、生物相互作用与中性过程如何共同塑造物种分布与群落结构, 是现代群落生态学的核心问题。然而, 传统多样性指数、排序分析及单物种分布模型(single-species distribution models, SDMs)难以同时整合物种间关联、环境梯度、性状与谱系等多维信息, 导致对群落构建机制的解析能力受限。联合物种分布模型(joint species distribution models, JSDMs)特别是生物群落层次建模(hierarchical modelling of species communities, HMSC)框架的提出, 为群落尺度的机制推断提供了统一而灵活的贝叶斯工具。本文系统综述了HMSC的统计结构、数学原理与推断机制, 构建了一个从数据组织、模型设定、马尔可夫链蒙特卡洛(Markov Chain Monte Carlo, MCMC)估计、模型评估到生态解释与预测的完整分析流程。同时结合苔藓群落数据配套编写了《联合物种分布模型HMSC的应用分步教程》, 通过分步讲解与可运行R代码, 助力研究者快速掌握该方法的实操应用。在理论部分, 本文明确了HMSC如何在统一的贝叶斯层级框架下整合环境梯度、物种性状、系统发育关系以及空间结构, 从而分离环境过滤、生物过滤与扩散限制的统计信号。在方法层面, 本文通过解析潜变量模型的数学结构, 阐明了残差相关在生态解释中的边界, 为理解物种共现信号、区分环境效应与未观测因子提供了理论依据; 对比了HMSC与其他主流JSDMs工具及传统群落统计方法的优势及适用性。在应用层面, 综述了其在森林、湿地、草原、海洋、城市及微生物生态学中的应用进展, 展示了其在保护规划、入侵种风险评估、共现网络分析及情景预测中的广泛价值; 随着图形处理器加速与迁移学习与大规模高维数据框架的发展, HMSC可提升稀有物种生态位估计与分布预测, 使数十万物种的群落建模成为可能。综上, JSDMs及HMSC不仅在生态统计方法论上实现了从单物种预测到多物种‒多维信息整合的跨越, 更为生态理论检验、群落构建机制解析及保护决策制定提供了高效、可扩展且能量化不确定性的工具平台。
表3
生物群落层次建模(HMSC)框架核心模型中的参数及其解释
正文中引用本图/表的段落
HMSC固定效应部分主要模拟环境过滤过程。核心模型参数的符号与解释详见表3。参数β描述物种特定的生态位, 能够量化环境变异对物种出现的影响; 参数Γ刻画响应性状对生态位的依赖性; 而当某些性状缺失且在系统发育上具有相关性时, 其生态位中的系统发育信号可通过ρ来表征。该部分不仅能够分离环境异质性对物种分布的贡献, 还能为识别性状-生态位关系及系统发育信号提供统计基础(Ovaskainen et al., 2017)。
HMSC的随机效应部分则用于刻画生物过滤与扩散限制。核心参数Ω表示在控制环境效应后的残差共现矩阵, 用以推断物种间的潜在相互作用; 参数α则反映无法由环境解释的空间变异尺度, 理论上与扩散过程相关(表3)。通过引入空间显式的随机效应, 模型可量化扩散限制对物种分布的影响。然而, 由于遗漏变量可能混入残差结构, 在解释Ω与α时需谨慎, 避免将其简单归因于生物相互作用或扩散过程(Ovaskainen & Abrego, 2020)。
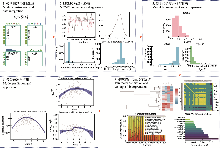
本教程以苔藓群落为研究示例, 在模型中同时考虑了多种生态因素: 温度、降水、土壤pH值和钙含量等连续环境梯度, 以及不同基质类型(如岩石、土壤、树皮、泥炭)等离散生境差异。模型还引入了苔藓的关键功能性状, 如持水能力、茎高、孢子大小和叶片厚度, 用来解释不同物种在环境变化下响应强度的差异。通过这些信息的结合, 模型能够从参数层面(如环境系数beta和性状调节系数gamma)揭示环境和性状的作用机制, 并在预测层面绘制出物种沿环境梯度的响应曲线以及群落层面的功能性状变化趋势, 从而把统计结果转化为生态机制的具体解释。此外, 模型特别加入了空间随机层, 用来捕捉地理上相邻样点之间的自然相似性, 防止环境变量的效应被空间聚集误判为“环境作用”。经过这一控制后, 残差相关仅表示在剔除环境和空间影响后的物种共变格局, 不能直接理解为物种间存在因果互作, 而是提示它们在微环境或未观测因子上的相似性。在方法学层面, 本教程涵盖了3种常见的数据类型与对应的似然结构: 出现-缺失数据使用二元probit模型, 计数数据采用泊松或负二项分布, 连续丰度数据则基于正态分布建模。针对苔藓群落数据, 教程提供了系统的模型诊断流程, 包括采样收敛与稳定性评估(比如有效样本量和潜在尺度缩减因子)以及出现-缺失模型的拟合优度检验(ROC曲线下面积与Tjur判别系数)。此外, 通过设置误设实验(如去除基质变量或关键性状), 教程演示了遗漏环境因子或性状信息时, 模型推断方向和强度可能出现的系统性偏差。在确保似然结构与随机层设置与数据类型相匹配, 并采用空间阻断式交叉验证以检验模型外推能力的前提下, 该建模框架具有广泛的适用性。它不仅适用于植物(包括苔藓、蕨类、裸子和被子植物), 也可推广至真菌与地衣群落、陆生动物(如昆虫、鸟类、两栖类、爬行类和哺乳类)、淡水与海洋生物(如鱼类、浮游生物、底栖无脊椎动物), 以及由细菌、古菌和真核生物构成的微生物群落。
在前述章节中, 我们对HMSC进行了系统介绍, 涵盖其数据结构、模型公式、先验设定与推断流程。HMSC作为一种高度整合的分层贝叶斯多物种分布模型, 不仅能够同时引入环境协变量、物种性状、系统发育信息以及空间-时间结构, 还可在统一框架下量化物种间的残差相关性, 因此在生态机制解析和多维数据融合方面具有显著优势。然而, JSDMs领域并不局限于HMSC一种实现, 近年来在推断引擎、计算性能、数据类型适应性及可扩展性上出现了多种各具特色的R包(Norberg et al., 2019), 如gllvm (Niku et al., 2019)、boral (Hui, 2016)、jSDM (Pollock et al., 2014)、sjSDM (Pichler & Hartig, 2021)和spOccupancy (Doser et al., 2022)等。它们在模型结构、数据维度支持、计算速度以及观测过程建模等方面存在差异, 导致在不同研究目的和数据条件下的适用性各不相同(附录1-表S3, 表S4)。
本文的其它图/表
|
备案号:京ICP备16067583号-7
Copyright © 2026 版权所有 《生物多样性》编辑部
地址: 北京香山南辛村20号, 邮编:100093
电话: 010-62836137, 62836665 E-mail: biodiversity@ibcas.ac.cn
![]()