



蚯蚓(环节动物门: 寡毛纲)作为一种古老的土壤无脊椎动物, 通过协调其他土壤生物促进生态系统多种功能服务(Liu T et al, 2019)。因为其强大的适应性及入侵性, 已经分布在世界各地(Phillips et al, 2019)。据统计, 2019年之前全世界共发现有3,000‒3,500种蚯蚓, 其中我国记录有640种, 是亚洲乃至全球报道蚯蚓物种数最为丰富的国家之一(Csuzdi, 2012; 蒋际宝和邱江平, 2018)。蚯蚓被喻为土壤中的“生态系统的工程师”, 其往往可以改变土壤的结构和质量, 目前已有大量研究表明蚯蚓在土壤中的摄食和挖掘习性对土壤微生物和植物的生长存在良性作用(Velki & Ecimovic, 2017; Medina-Sauza et al, 2019)。随着经济和社会的快速发展, 土壤污染情况日益严峻, 蚯蚓凭借其自身对有毒物质的敏感性和其他独特的生物学优势, 已经逐渐成为一种检测土壤健康的生物标志物(Shi et al, 2017; Li et al, 2020)。蚯蚓在土壤生态系统中独特的作用可以提高土壤中养分的利用效率, 同时也可以增强农作物对病虫害的抵抗力, 这也使其在农业中的应用日益广泛(Bertrand et al, 2015)。此外, 在国际上蚯蚓也被用来处理有机废弃物、净化污水等(Sinha et al, 2010; Bhat et al, 2018)。
人类对蚯蚓的研究历史非常悠久, 可追溯到1758年, 林奈描述并命名了第一个蚯蚓物种, 广泛分布在欧洲的陆正蚓(Lumbricus terrestris)。1892年, 达尔文在The Formation of Vegetable Mould, Through the Action of Worms: With Observations on Their Habits中曾说过: “我们很难找到其他的生灵像它们一样, 虽看似卑微, 却在世界历史的进程中起到了如此重要的作用” (Darwin, 1892)。虽然达尔文给予了蚯蚓高度的评价, 但目前对于蚯蚓的研究多集中于其分布、多样性、生态系统服务功能、生态毒理学、天然免疫, 如其体内蛋白质含量、激素分泌、药用价值以及生态习性等, 在基因组学方面的研究相对较少, 缺乏功能、机理的深入研究(Li et al, 2016; Ding et al, 2019; Jin et al, 2020; Parolini et al, 2020; Ahmed & Al-Mutairi, 2022), 使其在基因组、基因功能、生化、细胞、分子和发育等方面的研究远远落后于其他模式动物, 例如线虫等。
蚯蚓的物种繁多, 蚯蚓分类学仍然以性成熟个体的形态解剖方法为主, 但是由于大量蚯蚓幼体无显著的形态鉴定特征, 并且其基因的高度同源性导致分类鉴定仍有很长的路要走。从进化生物学的角度看, 蚯蚓有着丰富的形态特征(如无腿运动), 丰富的多样性, 进化保护等, 目前采用的分类鉴定方法主要有DNA条形码、多基因系统发育学、线粒体基因组分析、转录组分析、表达序列标签(expressed sequence tag, EST)等(Marchán et al, 2022)。但由于缺乏基因组的相关信息, 蚯蚓的鉴定及遗传进化等方面的研究也受到了一定限制(Marchán et al, 2020b; Rajesh & Anant, 2020)。如果可以丰富并完善其基因组学的相关研究, 蚯蚓将不仅限于作为土壤环境健康的指示生物, 而是逐渐向土壤模式动物的方向发展, 从而更为便捷地对环境进行指示服务(Shao et al, 2020), 并有可能揭示环境胁迫对蚯蚓个体和多样性影响的分子机制, 为探索蚯蚓适应土壤复杂多变环境的分子机制奠定基础。
近些年来, 关于蚯蚓基因组方面的研究虽也取得了一定进展, 但仅有4种蚯蚓的全基因组被测序完成, 测序蚯蚓种类和测序质量有待进一步提高, 目前多集中在全基因组的测序和组装, 以及基于线粒体基因组的分子系统发育等方面的研究, 缺少功能基因组的研究。据统计, 最早与蚯蚓基因组学相关的文献发表于1995年, 自2015起每年均有相关文章发表(图1)。本文基于Web of Science核心合集数据库, 以earthworm、genome为主题关键词进行论文检索, 筛选并对相关文献进行关键词分析后, 基于此使用VOSviewer构建并优化文献摘要共现图谱, 对现有的有关蚯蚓基因组的研究热点与趋势进行了分析(图2)。通过共现图谱可以发现, 目前的研究热点多集中在线粒体基因组、系统发育分析, 以此来为蚯蚓的物种多样性提供一定证据。同时对于爱胜蚓属(Eisenia)的研究相对较为丰富。本文总结了1995‒2022年Web of Science核心合集数据库现有蚯蚓基因组研究的成果, 并对今后蚯蚓基因组的研究方向提出了展望。
图1
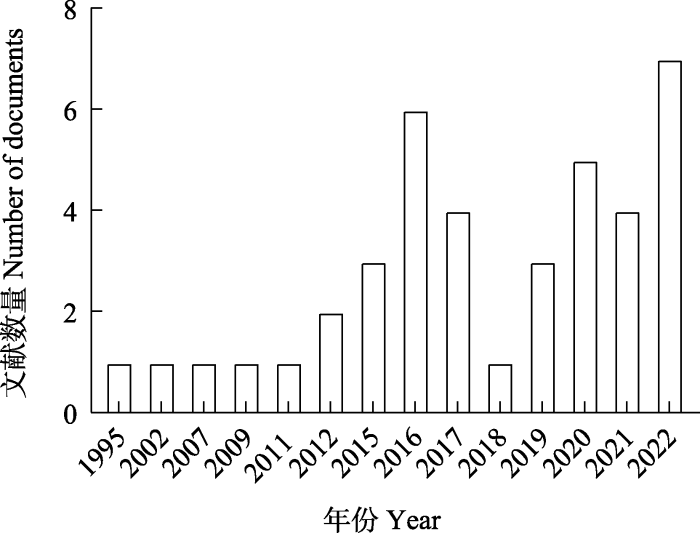
图1
蚯蚓基因组学相关文献发表数量。文献来源: Web of Science核心合集数据库; 时间: 1995‒2022年; 检索关键词: 蚯蚓、基因组。
Fig. 1
Number of published papers on earthworm genomics. Literature source: Web of Science Core Collection; Publication years: 1995‒2022; Search keywords: Earthworm, Genome.
图2
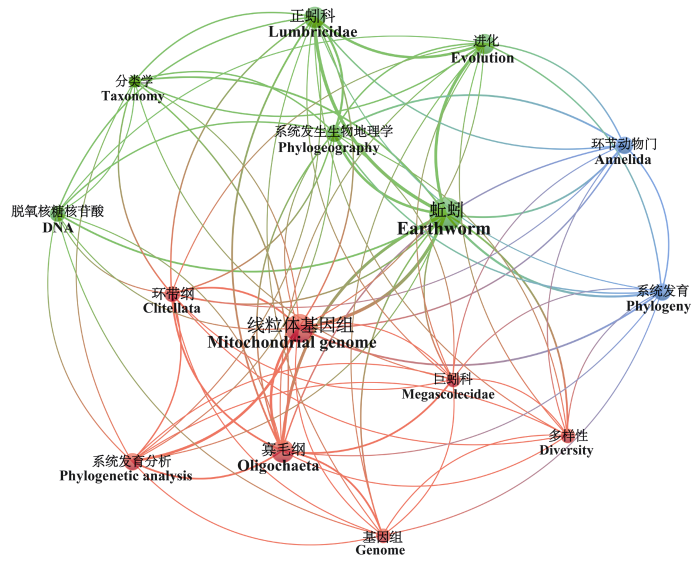
图2
蚯蚓基因组学研究关键词共现图谱
Fig. 2
Keywords coexistence network of research on earthworm genomics
1 蚯蚓全基因组研究进展
作为土壤动物的代表, 温度和降水量是影响蚯蚓全球分布最为关键的因素, 这也就使其广泛分布于除极地和干旱地区的全球各地, 而蚯蚓的挖洞、摄食和排泄等行为对维持土壤肥力至关重要(Pirooznia et al, 2007; Phillips et al, 2019)。蚯蚓在全球的广泛分布, 在一定程度上表明其在面对复杂多变的环境时具有极强的适应性, 这也就意味着蚯蚓往往具有强大的分子机制来维持自身的生存与繁衍(Wang et al, 2021)。这种强大的分子机制也逐渐引起了研究人员的重视, 但由于技术和资金等条件限制, 目前已知的有完整的全基因组信息的蚯蚓种类极少, 而对蚯蚓进行全基因组测序组装后, 编码蛋白质基因组注释、多基因组对比分析和系统发育研究可以为物种基因组进化提供更完整的见解(Jin et al, 2020)。并且, 通过全基因组测序分析还可以鉴别未知的蚯蚓, 例如不同蚓种的核基因组大小可能存在较大差异, Shekhovtsov等对诺登爱胜蚓(Eisenia nordenskioldi nordenskioldi)种组的小型(250‒500 Mbp)和大型(2,300‒3,500 Mbp)两组基因谱系进行了分析, 发现其祖先群体的基因组更可能为大型基因组(Marchán et al, 2020b; Shekhovtsov et al, 2021)。
此外, 在环境污染物胁迫下, 显著变化的蚯蚓基因、蛋白表达以及相关代谢物可作为分子生物标志物, 用于污染物的环境评价、胁迫检测、作用机制以及蚯蚓应对污染物的免疫防御机制的研究, 基于此目前已有大量针对蚯蚓的毒理研究。Gong和Perkins (2016)对已有的相关研究进行总结分析后发现, 目前已有多种生化、生理和分子等层面的蚯蚓生物标志物, 主要包括DNA损伤、DNA甲基化、金属结合蛋白(例如金属硫蛋白)的诱导、抗氧化系统的抑制、能量储备反应和免疫学变化、溶酶体膜稳定性、神经冲动传导性、精子数量和质量、组织病理学和行为等等, 并发现了相关基因存在显著变化, 但基因与蛋白质和代谢物之间的相互作用关系, 以及遗传和表观遗传变化在污染物及环境等胁迫条件的适应过程中的作用并不明确。
除了已知的关键基因, 在缺乏全基因组序列的情况下, 表达序列标签可以快速完成基因表达、基因功能和涉及特定生理过程的新基因鉴定, 是比较基因组和功能基因组研究的重要资源(Pirooznia et al, 2007; Liu et al, 2013)。在多种蚯蚓中, 赤子爱胜蚓(Eisenia foetida)常被用作土壤动物生态毒理的模式生物, 对其毒理分子机制的探究也是目前的研究热点之一。Pirooznia等人于2007年就已经完成了对赤子爱胜蚓进行基因功能注释, 分析其进化情况, 构建基因文库和对表达序列标签的序列分析, 可以完成表达基因鉴定并比较基因组差异, 进而更好地研究其对环境污染物的基因表达响应, 并且可以对关键基因(涉及生长、发育、再生、致病、免疫以及环境污染物扰动下的差异表达基因)进一步分析(Pirooznia et al, 2007; Zwarycz et al, 2015; Bhambri et al, 2018; Paul et al, 2018; Liu C et al, 2019)。全基因组测序技术虽然在快速发展, 但目前来看其成本依旧过高, 而简化基因组测序技术的出现与发展正在逐步弥补这一不足。
1.1 已测蚯蚓的全基因组信息
表1 蚯蚓全基因组测序基本信息
Table 1
| 蚯蚓 Earthworm | 基因组大小 Genome length | 染色体数量 Number of chromosomes | 染色体倍体 Chromosome ploidy | GC含量 GC content (%) | 蛋白编码基因数 Number of protein-coding gene | 测序组装技术 Sequencing assembly technology | 参考文献 References |
|---|---|---|---|---|---|---|---|
| 赤子爱胜蚓 Eisenia foetida | 1.05 Gb | 22 | 2 | 28.6 | ‒ | Illumina HiSeq 2000 2 × 100PE | 2015 |
| 赤子爱胜蚓 E. foetida | 1.47 Gb | 22 | 2 | 40 | ‒ | Illumina HiSeq 2500 | 2018 |
| 通俗腔蚓 Metaphire vulgaris | 728.6 Mb | 41 | 2 | 40 | ‒ | PromethION Single Molecule Platform, Illumina NovaSeq Sequencing Platform, Hi-C | 2020 |
| 安德爱胜蚓 Eisenia andrei | 1.3 Gb | 22 | 2 | 35‒45 | 31,817 | PacBio三代测序 PacBio RS Platform, Hi-C | 2020 |
| 皮质远盲蚓 Amynthas corticis | 1.2 Gb | 42 | 3 | 40.34 | 29,256 | PacBio三代测序 PacBio SMRT Sequencing, Hi-C | 2021 |
1.1.1 赤子爱胜蚓(Eisenia foetida)基因组解析
赤子爱胜蚓是目前常用的环境监测指示动物, 虽然其作为生态毒理研究模式生物是成功的, 但在全基因组研究之前仅有一组EST数据, 使其分子层面研究的发展受到了一定限制, 因此Zwarycz等(2015)对赤子爱胜蚓进行了全基因组测序与组装, 组装完成的结果为1.05 Gb, contig N50为1,850 bp。组装完成后对同源框进行了分类, 并与水蛭(Helobdella robusta)以及海蠕虫(Capitella teleta)进行比较后发现, 从赤子爱胜蚓与海蠕虫的最近共同祖先到赤子爱胜蚓与水蛭的最近共同祖先发生了许多基因扩增, 而从赤子爱胜蚓与水蛭的最近共同祖先到赤子爱胜蚓的谱系却具有异常水平的同源框增加, 为解释不同谱系的遗传多样性提供了一定的依据。随后, Bhambri等(2018)对赤子爱胜蚓进行了更完整的全基因组分析, 长度为1.47 Gb, contig N50为967 bp, 同时验证其二倍体染色体数目为22, GC含量达到40%, 更加完备的全基因组序列的分析为研究赤子爱胜蚓的发育多样性的遗传原因提供了重要工具。
1.1.2 安德爱胜蚓(Eisenia Andrei)基因组解析
除了在环境方面的应用, 蚯蚓的再生过程也是研究人员的关注点之一。再生是最为复杂的生物学过程之一, 蚯蚓以其自身复杂的表型结构, 较短的再生周期以及可以双向再生等优势, 表明其可以作为一种研究再生机制的优秀动物模型。Shao等(2020)利用PacBio测序和Hi-C辅助组装等策略拼装了一个染色体水平的高质量安德爱胜蚓基因组, Scaffold N50约为111 Mb, 并结合蚯蚓不同再生时期转录组和单细胞转录组测序对再生发育机制进行了探究, 发现典型的再生通路Wnt信号通路在安德爱胜蚓显著扩张, 蚯蚓基因组中重复序列LINE2转座元件以及与再生功能相关的基因家族(例如, EGFR1, 表皮生长因子受体)也有显著的扩增, 这些显著扩增的基因家族可能通过增加其拷贝数剂量效应来调控蚯蚓再生过程, 这为蚯蚓再生提供关键基因。
1.1.3 通俗腔蚓(Metaphire vulgaris)基因组解析
Jin等(2020)对通俗腔蚓的全基因进行了测序分析、组装与注释, 组装的序列由559个重叠群组成, 长度为728.6 Mb, N50重叠群大小为4.2 Mb。根据Hi-C结果, 重叠群锚定在41条染色体上。基因组序列核基因比较表明通俗腔蚓发生了全基因组重复, 随后发生了几次染色体融合事件。Hox基因和蚓激酶基因为基因组周围的部分簇。揭示了从古代四倍体到现代二倍体物种的进化路线, 基因组复制(四倍体化)可能导致通俗腔蚓体型更大。同时该研究发现Hox基因和蚓激酶基因是基因组周围的部分簇,通俗腔蚓基因组的测序和组装为蚯蚓的基因功能和进化研究提供参考。
1.1.4 皮质远盲蚓(Amynthas cortices)基因组解析
皮质远盲蚓具有强大适应能力, 现已广泛分布于世界各地。Wang等(2021)基于长读段三代测序与Hi-C测序相结合的策略, 对蚯蚓基因组进行了高质量拼装, 形成了一个长达1.2 Gb, 包含42条准染色体序列的基因组。蚯蚓基因组拼装序列的N50长度达到31 Mb, 结合转录组测序数据共预测出29,256个蛋白质编码基因, 且91.2%的后生动物单拷贝直系同源基因都在该基因组中存在完整匹配, 证明该基因组具有较高的完整性。这为深入探究隐藏在蚯蚓物种独特性背后的分子机制提供了先决条件。核型分析发现皮质远盲蚓具有多于120条的染色体, 通过杂合度估计的K-mer分析、倍性估计和核型分析, 发现其为三倍体物种, 这也就意味着其会有更多的基因材料用于适应性进化, 并且应激/防御基因会有更多的拷贝。同时, 与赤子爱胜蚓或安德爱胜蚓相比, 皮质远盲蚓基因组中与免疫系统过程、应激反应和稳态过程相关的基因功能更加丰富。这也就产生了更多的防御功能, 该研究通过对蚯蚓基因组的完整拼装与多组学分析, 揭示了世界广布种皮质远盲蚓基因组的三倍体特征及其与肠道微生物、土壤紧密互作的分子机制, 并为后续研究蚯蚓的适应机制提供了丰富且多元的数据资源。
赤子爱胜蚓与安德爱胜蚓为近缘物种, 两者同样为二倍体, 染色体数目相同, 但安德爱胜蚓基因组最终组装的大小为1.3 Gb, 略小于赤子爱胜蚓。安德爱胜蚓使用的是PacBio三代测序和Hi-C测序, 赤子爱胜蚓用的是Illumina HiSeq 2500二代测序, 相比较来说安德爱胜蚓的基因组测序核组装质量要高于赤子爱胜蚓, 使用BUSCO验证后, 赤子爱胜蚓基因组中的识别结果为18.2%, 而安德爱胜蚓则高达92.1%。关于两者的基因组学研究均表明, 基因家族的扩增或收缩会与特定表型和生理功能的进化有关, 并且在两者的基因组中均发现了丰富的扩展基因家族, 而这些基因复制发生在赤子爱胜蚓与安德爱胜蚓分化之前(Zwarycz et al, 2015; Bhambri et al, 2018; Shao et al, 2020)。已测定的通俗腔蚓与皮质远盲蚓同为巨蚓科, 通俗腔蚓为二倍体, 皮质远盲蚓则为三倍体, 皮质远盲蚓测序质量高于通俗腔蚓。
1.2 简化基因组测序(reduced-representation genome sequencing, RRGS)
由于全基因组测序对于资金和技术的要求较高, 尤其是对于基因组偏大的物种, 而研究通常都想要覆盖全基因组的变异信息, 这就催生了简化基因测序技术, 这种技术在没有参考基因组的物种中也可以使用, 从而可以帮助人们进行生物系统发育分析、物种鉴定以及种群进化等方面的研究(Yuan et al, 2020; Marchán et al, 2020b)。限制性酶切位点关联DNA测序(restriction site-associated DNA sequencing, RAD-seq)以及基因分型测序(genotyping- by-sequencing, GBS)是目前使用最为广泛的简化基因组测序技术(Andrews et al, 2016; Marchán et al, 2020a)。
蚯蚓由于其强大的环境适应力, 以及庞大的种群数量, 常被认为是高度多态性的动物, 而它的多态性不仅仅体现在其形态层面的多样性, 更存在于其基因层面, 因此近些年来对于蚯蚓种群遗传变异的研究也日渐丰富(Yuan et al, 2020)。全基因组单核苷酸多态性(single nucleotide polymorphism, SNP)的发现对于遗传图谱的研究至关重要, 并且SNPs目前仍广泛应用于蚯蚓的各项研究(Baird et al, 2008; Marchán et al, 2020a)。Anderson等(2017)则通过RAD-seq对生存在高度污染的前矿区的粉正蚓(Lumbricus rubellus)的基因组进行了分析, 并与附近正常地区的蚯蚓基因组进行比较, 证明谱系之间不存在混合, 发现许多SNPs分离了种群, 表明这些基因被选择下来应对重金属毒性。Yuan等(2020)利用RAD-seq技术, 使用SNP标记揭示了巨蚓科蚯蚓Amynthas_YN2017 sp.的群体遗传分化和遗传结构, 并推测地理隔离导致的基因流减少影响了Amynthas_YN2017 sp.的群体遗传分化, 该研究也表明RAD-seq技术可以用于探索蚯蚓的种群系统发育关系和遗传多样性。
以上研究为蚯蚓的遗传研究、基因克隆和系统发育分析、免疫防御等提供了宝贵资源。蚯蚓的全基因组测序为同源类群比较、系统发育、基因组筛选注释预测编码基因序列, 以及了解基因组进化和多样性变化提供了关键资源。
2 基于线粒体基因组分析的蚯蚓分类学和系统发育研究
经典的蚯蚓分类主要是基于形态结构的比较。目前对于成年蚯蚓的鉴定, 在形态层面主要基于体色、体长、前列腺类型、刚毛位置和多寡、阴蒂位置和形式、环带位置及形状以及一些内脏器官, 如精巢囊和受精囊等(Pérez-Losada et al, 2009)。但相比于节肢动物, 蚯蚓可用于分类的性状依旧是极少的, 这可能与蚯蚓的世系始终在土壤中演化有关(树蚓例外)。虽然土壤密实和黑暗的环境很可能在一定程度上限制了蚯蚓形态的分化, 但由于蚯蚓的某些性状(如体色)可塑性较强, 不同属或物种在同一特征中也可能会表现出相同性状。同时由于个体部分特征差异较大, 还有些形态特征为可变的, 这就对蚓种的分类及鉴定工作造成了一定的障碍(Pérez-Losada et al, 2009)。
在严苛的土壤环境中, 形态分化极为缓慢或可能停滞, 但是DNA的变异不会停滞, 近些年随着分子技术的快速发展, 分子数据为蚯蚓的分类带来了动能。21世纪初, 利用Sanger测序获得单基因或寡基因数据, 通过分子系统学手段为蚯蚓分类学、系统发育学和生物地理学研究提供了更为可靠的结果(Bozorgi et al, 2019)。蚯蚓在分子系统发育学最早的研究突破之一是DNA条形码的实现, 如细胞色素C氧化酶亚基I序列(COX1), 其他线粒体基因(COX2、ND1、12S与16S)以及核基因分子标记(28S、H3)等。这一方法可以帮助破译蚯蚓的不同扩散和定居模式, 以及它们的功能特征、地质历史和人为影响的潜在关系。分子系统学研究也主要集中在基于单基因或联合基因片段的系统分类与谱系地理学方面, 比如: 蚯蚓隐存种(cryptic species)的代表类群, 基于核基因与线粒体基因片段的背暗流蚓种组(Aporrectodea caliginosa species-complex)的系统发育以及地中海优势种Hormogaster elisae species-complex的谱系地理学与遗传景观分析(Pérez-Losada et al, 2009; Marchán et al, 2017)。基于多基因片段, 鉴定出伊朗波斯湾地区正蚓科3个新种, 并揭示了1个新属Philomontanus (Bozorgi et al, 2019)。
表2 环带纲蚯蚓线粒体基因组测序物种的分类单元分布
Table 2
| 分类单元 Taxa | GenBank登录号 GenBank accession no. | 参考文献 References |
|---|---|---|
| 正蚓科 Lumbricidae | ||
| Aporrectodea caliginosa | CM035405.1 | 未发表 Unpublished |
| A. rosea | NC_046733.1 | 2019 |
| A. tuberculata | OL840316−OL840317, OM687883−OM687886 | 2022 |
| A. trapezoides | OM687887 | 2022 |
| Bimastos parvus | MZ857199.1 | 2021 |
| Dendrobaena octaedra | MZ857197.1 | 2021 |
| Eisenia andrei | MZ857198.1 | 2021 |
| E. nordenskioldi | MZ857200.1 OL840314−OL840315 OM687887−OM687890 | 2021 2022 2022 |
| Lumbricus rubellus | MN102127.1 | 2019 |
| L. terrestris | NC_001673.1 | 1995 |
| Octolasion tyrtaeum | MZ857201.1 | 2021 |
| Pontoscolex corethrurus | NC_034783.1 | 未发表 Unpublished |
| 巨蚓科 Megascolecidae | ||
| Amynthas aspergillus | NC_025292.1 | 2016b |
| A. carnosus | KT429008.1 | 2016c |
| A. corticis | KM199290.1 | 2015 |
| A. cucullatus | KT429012.1 | 2016c |
| A. gracilis | KP688582.1 | 2015 |
| A. hupeiensis | KT429009.1 | 2016c |
| A. instabilis | KT429007.1 | 2016c |
| A. jiriensis | NC_029879.1 | 2017 |
| A. longisiphonus | KM199289.1 | 2015 |
| A. moniliatus | KT429020.1 | 2016c |
| A. morrisi | KT429011.1 | 2016c |
| A. pectiniferus | KT429018.1 | 2016c |
| A. redactus | KT429010.1 | 2016c |
| A. robustus | KT429019.1 | 2016c |
| A. rongshuiensis | KT429014.1 | 2016c |
| A. seungpanensis | OL321943.1 | 未发表 Unpublished |
| A. spatiosus | KT429013.1 | 2016c |
| A. triastriatus | KT429016.1 | 2016c |
| A. yunoshimensis | LC573969.1 | 2021 |
| Duplodicodrilus schmardae | KT429015.1 | 2016c |
| Metaphire californica | KP688581.1 | 2015 |
| M. guillelmi | KT429017.1 | 2016c |
| M. hilgendorfi | LC573968.1 | 2021 |
| M. vulgaris | NC_023836.1 | 2016a |
| Perionyx excavatus | NC_009631.1 | 未发表 Unpublished |
| Tonoscolex birmanicus | KF425518.1 | 2015 |
| 链胃蚓科 Moniligastridae | ||
| Drawida gisti | NC_058285.1 | 2020 |
| D. japonica | KM199288.1 | 2016c |
| D. ghilarovi | OL840312−OL840313 | 2022 |
2.1 蚯蚓线粒体基因组的特征
蚯蚓线粒体组为双链环状DNA, 包含37个基因, 即13个蛋白编码基因、22个转运RNA和2个核糖体RNA, 有些物种的线粒体组里包含控制区(control region), 有些物种则没有。蚯蚓线粒体组区别于大多数无脊椎动物和所有脊椎动物的显著特征是所有37个基因都位于正链上(图3), 而其他动物都是正负链均有分布, 表明线粒体基因顺序在寡毛纲动物中始终是保守的(Zhang et al, 2016c; Zhao et al, 2022)。目前报道的多数蚯蚓的线粒体组都是不完整的, 可能的原因是控制区的部分重复序列无法通过二代测序获得, 因为二代测序的reads一般都比较短(150−400 bp)无法测通长片段重复区域, 需要结合Sanger扩增控制区来重建环状DNA, 或者直接使用三代测序手段获得线粒体组的完整序列。
图3
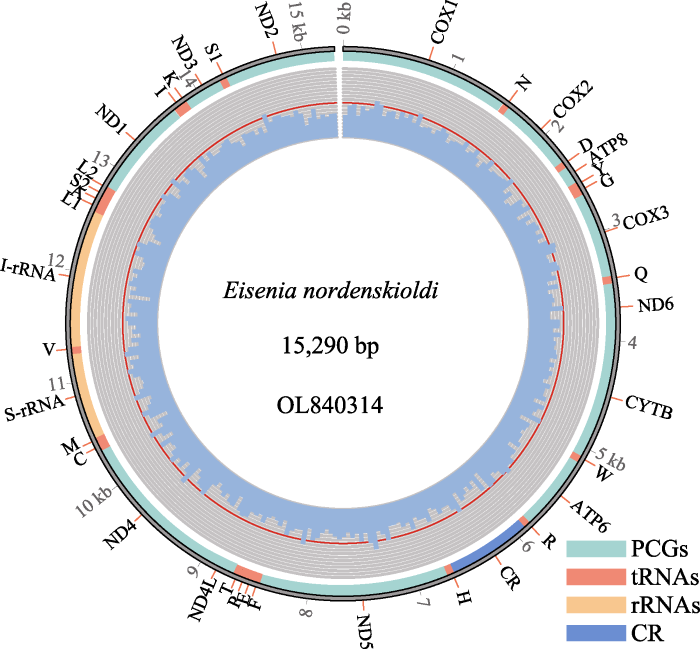
图3
诺登爱胜蚓的线粒体基因组。青色代表蛋白编码基因, 红色代表转运RNA, 橙色代表核糖体RNA, 蓝色代表控制区。
Fig. 3
The mitochondrial genome of Eisenia nordenskioldi. Cyan color represents protein-coding genes, red color represents transporter RNA, orange color represents ribosomal RNA, and blue color represents the control region.
2.2 蚯蚓线粒体基因组的应用
随着线粒体基因组分析方法的普及, 目前, 蚯蚓线粒体组的数据主要用于分类学、系统发育和谱系地理。诺登爱胜蚓作为研究较多的蚓种之一, 其广泛分布于北亚及邻近地区, 以其核型和遗传变异而闻名, Zhang等(2022)对中国雾灵山地区的诺登爱胜蚓种组进行采样分析后发现, 海拔会影响其遗传多样性, 但不影响其外部形态。同时诺登爱胜蚓也常被认为是最近入侵欧洲的物种, 但Shekhovtsov等(2016, 2020)通过对其线粒体基因组进行分析后发现了其在欧洲的另一遗传谱系, 且亲缘关系较远, 并通过分子时钟推测, 这两种谱系在末次盛冰期就已出现, 因此提出了诺登爱胜蚓在欧洲并不是入侵物种的观点, 而这与传统观点是相悖的。而且诺登爱胜蚓极有可能由西伯利亚经蒙古扩散至河北北部山区, 并在海拔梯度下形成了特有的遗传谱系(Zhang et al, 2022)。对粉正蚓(Lumbricus rubellus)的线粒体基因组分析发现同地域不同谱系的粉正蚓差异较大, 但并不存在生殖隔离, 这也就说明粉正蚓是一种高度多态的物种, 而不是常规认为的是几种未鉴定蚓种的种组(Giska et al, 2015; Zhang et al, 2019)。
3 展望
随着现代生物信息学技术的不断发展, 分子机制的研究也逐渐与基因组学、转录组学、蛋白组学、代谢组学等息息相关。而对蚯蚓来说, 不管是利用毒理基因组学挖掘新的生物标志物达到完善其环境监测功能的目的, 还是对其免疫防御机制探究, 都因缺乏基因组的信息而受到一定的限制, 这也就是为什么蚯蚓的研究多停留在观察、描述、生理等层面。目前已经有越来越多的研究人员参与完善蚯蚓的基因组信息, 并在不断地进行方法创新, 蚯蚓基因组测序的完成标志着人类对于蚯蚓生命现象和过程有了进一步的认识。
3.1 全基因组分析
目前完成全基因测序的蚯蚓仅有4种(Amynthas corticis、Eisenia andrei、E. foetida、Metaphire vulgaris), 这4种蚯蚓的全基因组信息已相对完善, 并且后续相关研究已逐步展开。但相对于蚯蚓庞大的种群数量来说, 这是微不足道的, 所以后续需要对更多的蚯蚓种进行全基因组测序。
全基因组学分析可以更为全面且准确地鉴别蚓种、分析遗传进化等, 在今后的研究中, 可以就已有的全基因组信息进行更深层次的挖掘, 例如与蚯蚓生长发育、免疫防御相关的基因功能, 这将会从本质上理解蚯蚓的生态服务功能、如何发挥环境指示动物的作用以及回答蚯蚓适应土壤复杂环境的分子机制。根据蚯蚓基因组信息发现新的功能基因, 研发蚯蚓功能蛋白, 利用基因工程技术, 体外生产蚯蚓功能蛋白, 用于医药、功能蛋白和饲料等方面。
3.2 线粒体基因组分析
相对于全基因组信息的缺乏, 现已有多种蚯蚓的线粒体基因组已经被测定并分析, 但对于现有的蚓种数量来说是微不足道的, 因此线粒体基因组的测序工作任重道远。蚯蚓现已广泛分布于全球, 且蚓种间存在生殖隔离, 但对其进化过程的研究并不明确, 因此对于已有的线粒体基因组信息, 可以从蚯蚓的地域分布与进化过程两个角度进行更深入的对比与分析, 这将更好地对蚯蚓进行更好的应用与保护。
综上, 根据目前的研究进展, 本文建议未来可以从以下几个方面开展对蚯蚓基因组学的深入研究: (1)针对现有的4种蚯蚓全基因组测序结果, 进一步从比较基因组学、进化基因组学和功能基因组学的方向分析, 从而能够更加深入理解蚯蚓的多样化、环境适应的分子机制以及种群进化机制。(2)完善不同种蚯蚓的基因文库和表达序列标签, 目前在这方面信息完善的蚯蚓种类较少, 这也在一定程度上限制了蚯蚓分子技术的发展。(3)加强线粒体基因组以及核基因组的综合分析, 力求解决更高层次的蚯蚓系统发育问题。蚯蚓基因组学研究才刚刚开始, 并且存在诸多困难, 但这是未来蚯蚓研究的必经之路。
参考文献
Earthworms effect on microbial population and soil fertility as well as their interaction with agriculture practices
DOI:10.3390/su14137803 URL [本文引用: 1]
Genetic variation in populations of the earthworm, Lumbricus rubellus, across contaminated mine sites
DOI:10.1186/s12863-017-0557-8
PMID:29149838
[本文引用: 1]

Background: Populations of the earthworm, Lumbricus rubellus, are commonly found across highly contaminated former mine sites and are considered to have under-gone selection for mitigating metal toxicity. Comparison of adapted populations with those found on less contaminated soils can provide insights into ecological processes that demonstrate the long-term effects of soil contamination. Contemporary sequencing methods allow for portrayal of demographic inferences and highlight genetic variation indicative of selection at specific genes. Furthermore, the occurrence of L. rubellus lineages across the UK allows for inferences of mechanisms associated with drivers of speciation and local adaptation.Results: Using RADseq, we were able to define population structure between the two lineages through the use of draft genomes for each, demonstrating an absence of admixture between lineages and that populations over extensive geographic distances form discrete populations. Between the two British lineages, we were able to provide evidence for selection near to genes associated with epigenetic and morphological functions, as well as near a gene encoding a pheromone. Earthworms inhabiting highly contaminated soils bare close genomic resemblance to those from proximal control soils. We were able to define a number of SNPs that largely segregate populations and are indicative of genes that are likely under selection for managing metal toxicity. This includes calcium and phosphatehandling mechanisms linked to lead and arsenic contaminants, respectively, while we also observed evidence for glutathione-related mechanisms, including metallothionein, across multiple populations. Population genomic end points demonstrate no consistent reduction in nucleotide diversity, or increase in inbreeding coefficient, relative to history of exposure.Conclusions: Though we can clearly define lineage membership using genomic markers, as well as population structure between geographic localities, it is difficult to resolve markers that segregate entirely between populations in response to soil metal concentrations. This may represent a highly variable series of traits in response to the heterogenous nature of the soil environment, but ultimately demonstrates the maintenance of lineage-specific genetic variation among local populations. L. rubellus appears to provide an exemplary system for exploring drivers for speciation, with a continuum of lineages coexisting across continental Europe, while distinct lineages exist in isolation throughout the UK.
Harnessing the power of RADseq for ecological and evolutionary genomics
DOI:10.1038/nrg.2015.28
PMID:26729255
[本文引用: 1]
High-throughput techniques based on restriction site-associated DNA sequencing (RADseq) are enabling the low-cost discovery and genotyping of thousands of genetic markers for any species, including non-model organisms, which is revolutionizing ecological, evolutionary and conservation genetics. Technical differences among these methods lead to important considerations for all steps of genomics studies, from the specific scientific questions that can be addressed, and the costs of library preparation and sequencing, to the types of bias and error inherent in the resulting data. In this Review, we provide a comprehensive discussion of RADseq methods to aid researchers in choosing among the many different approaches and avoiding erroneous scientific conclusions from RADseq data, a problem that has plagued other genetic marker types in the past.
Earthworm services for cropping systems. A review
DOI:10.1007/s13593-014-0269-7 URL [本文引用: 1]
Earthworms as organic waste managers and biofertilizer producers
DOI:10.1007/s12649-017-9899-8 URL [本文引用: 1]
Complete sequence of the mitochondrial DNA of the annelid worm Lumbricus terrestris
DOI:10.1093/genetics/141.1.305
PMID:8536978
[本文引用: 1]
We have determined the complete nucleotide (nt) sequence of the mitochondrial genome of an oligochaete annelid, the earthworm Lumbricus terrestris. This genome contains the 37 genes typical of metazoan mitochondrial DNA (mtDNA), including ATPase8, which is missing from some invertebrate mtDNAs. ATPase8 is not immediately upstream of ATPase6, a condition found previously only in the mtDNA of snails. All genes are transcribed from the same DNA strand. The largest noncoding region is 384 nt and is characterized by several homopolymer runs, a tract of alternating TA pairs, and potential secondary structures. All protein-encoding genes either overlap the adjacent downstream gene or end at an abbreviated stop codon. In Lumbricus mitochondria, the variation of the genetic code that is typical of most invertebrate mitochondrial genomes is used. Only the codon ATG is used for translation initiation. Lumbricus mtDNA is A + T rich, which appears to affect the codon usage pattern. The DHU arm appears to be unpaired not only in tRNAser(AGN), as is typical for metazoans, but perhaps also in tRNAser(UCN), a condition found previously only in a chiton and among nematodes. Relating the Lumbricus gene organization to those of other major protostome groups requires numerous rearrangements.
The complete mitochondrial DNA sequence of the pantropical earthworm Pontoscolex corethrurus (Rhinodrilidae, Clitellata): Mitogenome characterization and phylogenetic positioning
DOI:10.3897/zookeys.688.13721 URL [本文引用: 2]
Earthworms: A source of protein
Deeply divergent sympatric mitochondrial lineages of the earthworm Lumbricus rubellus are not reproductively isolated
DOI:10.1186/s12862-015-0488-9 URL [本文引用: 1]
Earthworm toxicogenomics: A renewed genome-wide quest for novel biomarkers and mechanistic insights
DOI:10.1016/j.apsoil.2015.11.005 URL [本文引用: 1]
Complete mitochondrial genome of the earthworm
Origin and evolution of earthworms belonging to the family Megascolecidae in China
DOI:10.17520/biods.2018105
[本文引用: 1]
A total of 579 Megascolecidae species have been reported in China through 2017. Most belong to the genera Amynthas and Metaphire. The family’s rich diversity merits further investigation into its evolutionary history. The arc of research indicates that analysis based on taxonomical and molecular methods reveals the evolutionary history of the family Megascolecidae in China. This paper summarizes updated findings regarding origin, speciation and dispersal of Megascolecidae in China, and discusses the evolution of major familial characteristics (e. g. spermathecal pores, caeca). Several analyses suggest that the ancestors of Megascolecidae in China may have come from the Indo-China Peninsula. Speciation radiation likely occurred after the Cretaceous-Palaeogene mass extinction, and species richness increased rapidly during the Cenozoic period. Ancestral range reconstruction analysis shows that species-groups are polyphyletic and that evolutionary reversal often resulted in sharp evolution of caeca, so we suggest that the current taxonomic system of Megascolecidae, which is based on a few morphological characters, should be reconstructed. We also note that the specific mechanism of evolution in Megascolecidae has not been studied. Hence, future research to reveal the specific evolutionary mechanism of Megascolecidae earthworms requires more systematic sampling of this family, combined with morphological research, phylogeny construction and analysis of geographical patterns, geological history and environmental factors.
中国巨蚓科蚯蚓的起源与演化
DOI:10.17520/biods.2018105
[本文引用: 1]
截至2017年中国已记录巨蚓科蚯蚓579种(亚种), 优势类群为远盲蚓属(Amynthas)与腔蚓属(Metaphire), 其丰富的物种多样性值得深入研究。近年来的研究指出联合形态分类与分子系统发育可较好地探讨中国巨蚓科蚯蚓的起源、分化与扩散。本文概述了中国巨蚓科蚯蚓物种的起源、分化时间以及扩散历程, 探讨了受精囊孔、盲肠形状等重要特征的演化, 分析了现行分类系统的缺陷。多项研究表明, 中国巨蚓科主要类群的祖先起源于中南半岛, 于白垩纪末期至新生代初期进入中国, 在新生代得以繁荣发展; 受精囊孔的对数或位置是多起源的, 盲肠形状演化中有祖征重现的现象, 故使用少数形态特征进行类群划分的现行分类系统有待改进。此外, 中国巨蚓科蚯蚓的具体演化机制及影响因素仍不明确。因此, 在未来研究中整合形态特征、分子数据与地理格局、地质历史及环境因子等信息, 定量分析类群演化与古地理、生物与非生物因素间的关系, 将有助于全面厘清中国巨蚓科蚯蚓演化的具体机制。
High-quality genome assembly of Metaphire vulgaris
Enrofloxacin at environmentally relevant concentrations enhances uptake and toxicity of cadmium in the earthworm Eisenia fetida in farm soils
DOI:10.1016/j.jhazmat.2016.01.057 URL [本文引用: 1]
Ecotoxicological effects of petroleum-contaminated soil on the earthworm Eisenia fetida
DOI:10.1016/j.jhazmat.2020.122384 URL [本文引用: 1]
Construction of a cDNA library and preliminary analysis of the expressed sequence tags of the earthworm Eisenia fetida (Savigny, 1826)
Construction of a full-length enriched cDNA library and preliminary analysis of expressed sequence tags from Bengal tiger Panthera Tigris Tigris
DOI:10.3390/ijms140611072 URL [本文引用: 1]
Characterization of five new earthworm mitogenomes (Oligochaeta: Lumbricidae): Mitochondrial phylogeny of Lumbricidae
DOI:10.3390/d13110580 URL
Earthworms coordinate soil biota to improve multiple ecosystem functions
DOI:S0960-9822(19)31095-4
PMID:31587999
[本文引用: 1]
Earthworms have been perceived as benevolent soil engineers since the time of Charles Darwin, but several recent syntheses link earthworm activities to higher greenhouse gas emissions, less soil biodiversity, and inferior plant defense against pests. Our study provides new field-based evidence of the multiple direct and indirect impacts of earthworms on ecosystem functions within an ecological multifunctionality framework (i.e., aggregated measures of the ability of ecosystems to simultaneously provide multiple ecosystem functions). Data from a 13-year field experiment describing 21 ecosystem functions showed that earthworm presence generally enhanced multifunctionality by indirect rather than direct effects. Specifically, earthworms enhanced multifunctionality by shifting the functional composition toward a soil community favoring the bacterial energy channel and strengthening the biotic associations of soil microbial and microfaunal communities. However, earthworm-mediated changes in soil physical structure, pH, and taxonomic diversity were not related to multifunctionality. We conclude that the coordinated actions of earthworms and their associated soil biota were responsible for the maintenance of multifunctionality at high levels in this rice-wheat cropping system. Management of crop residue inputs and reduction of soil physicochemical disturbances should encourage beneficial earthworm effects and support multiple ecosystem services that are vital to sustainable agriculture.Copyright © 2019 Elsevier Ltd. All rights reserved.
Perspectives in earthworm molecular phylogeny: Recent advances in Lumbricoidea and standing questions
DOI:10.3390/d14010030 URL [本文引用: 2]
Pinpointing cryptic borders: Fine-scale phylogeography and genetic landscape analysis of the Hormogaster elisae complex (Oligochaeta, Hormogastridae)
DOI:S1055-7903(17)30080-5
PMID:28487260
[本文引用: 2]
Spatial and temporal aspects of the evolution of cryptic species complexes have received less attention than species delimitation within them. The phylogeography of the cryptic complex Hormogaster elisae (Oligochaeta, Hormogastridae) lacks knowledge on several aspects, including the small-scale distribution of its lineages or the palaeogeographic context of their diversification. To shed light on these topics, a dense specimen collection was performed in the center of the Iberian Peninsula - resulting in 28 new H. elisae collecting points, some of them as close as 760m from each other- for a higher resolution of the distribution of the cryptic lineages and the relationships between the populations. Seven molecular regions were amplified: mitochondrial subunit 1 of cytochrome c oxidase (COI), 16S rRNA and tRNA Leu, Ala, and Ser (16S t-RNAs), one nuclear ribosomal gene (a fragment of 28S rRNA) and one nuclear protein-encoding gene (histone H3) in order to infer their phylogenetic relationships. Different representation methods of the pairwise divergence in the cytochrome oxidase I sequence (heatmap and genetic landscape graphs) were used to visualize the genetic structure of H. elisae. A nested approach sensu Mairal et al. (2015) (connecting the evolutionary rates of two datasets of different taxonomic coverage) was used to obtain one approximation to a time-calibrated phylogenetic tree based on external Clitellata fossils and a wide molecular dataset. Our results indicate that limited active dispersal ability and ecological or biotic barriers could explain the isolation of the different cryptic lineages, which never co-occur. Rare events of long distance dispersal through hydrochory appear as one of the possible causes of range expansion.Copyright © 2017 Elsevier Inc. All rights reserved.
Genome-informed integrative taxonomic description of three cryptic species in the earthworm genus Carpetania (Oligochaeta, Hormogastridae)
DOI:10.1080/14772000.2020.1730474 URL [本文引用: 3]
Local adaptation fuels cryptic speciation in terrestrial annelids
DOI:10.1016/j.ympev.2020.106767 URL [本文引用: 3]
Earthworms building up soil microbiota, a review
DOI:10.3389/fenvs.2019.00081 URL [本文引用: 1]
Earthworm as an alternative protein source in poultry and fish farming: Current applications and future perspectives
DOI:10.1016/j.scitotenv.2020.139460 URL [本文引用: 1]
Data on genome annotation and analysis of earthworm Eisenia fetida
DOI:10.1016/j.dib.2018.08.067 URL [本文引用: 1]
Phylogenetic assessment of the earthworm Aporrectodea caliginosa species complex (Oligochaeta: Lumbricidae) based on mitochondrial and nuclear DNA sequences
DOI:10.1016/j.ympev.2009.04.003
PMID:19364539
[本文引用: 3]
The Aporrectodea caliginosa species complex includes the most abundant earthworms in grasslands and agricultural ecosystems of the Paleartic region. Historically this complex consisted of the following taxa: A. caliginosa s.s.Savigny, 1826, A. trapezoides Dugés (1828), A. tuberculata (Eisen, 1874), and A. nocturna Evans (1946). These four taxa are morphologically very similar and difficult to differentiate because of their morphological variability. Consequently, their taxonomic status and their phylogenetic relationships have been a matter of discussion for more than a century. To study these questions, we sequenced the COII (686 bp), 12S (362 bp), 16S (1200 bp), ND1 (917 bp), and tRNAs(Asn-Asp-Val-Leu-Ala-Ser-Leu) (402 bp) mitochondrial and 28S (809 bp) nuclear gene regions for 85 European earthworms from 27 different localities belonging to the A. caliginosa species complex and four outgroup taxa. DNA sequences were analyzed using maximum parsimony, maximum likelihood, and Bayesian approaches of phylogenetic inference. The resulting trees were combined with morphological, ecological, and genomic evidence to test species boundaries (i.e., integrative approach). Our molecular analyses showed that A. caliginosa s.s. and A. tuberculata form a sister clade to A. trapezoides, A. longa, and A. nocturna, which indicates that A. longa is part of the A. caliginosa species complex. We confirm the species status of all these taxa and identify two hitherto unrecognized Aporrectodea species in Corsica (France). Moreover our analyses also showed the presence of highly divergent lineages within A. caliginosa, A. trapezoides, and A. longa, suggesting the existence of cryptic diversity within these taxa.
Global distribution of earthworm diversity
DOI:10.1126/science.aax4851
PMID:31649197
[本文引用: 2]
Soil organisms, including earthworms, are a key component of terrestrial ecosystems. However, little is known about their diversity, their distribution, and the threats affecting them. We compiled a global dataset of sampled earthworm communities from 6928 sites in 57 countries as a basis for predicting patterns in earthworm diversity, abundance, and biomass. We found that local species richness and abundance typically peaked at higher latitudes, displaying patterns opposite to those observed in aboveground organisms. However, high species dissimilarity across tropical locations may cause diversity across the entirety of the tropics to be higher than elsewhere. Climate variables were found to be more important in shaping earthworm communities than soil properties or habitat cover. These findings suggest that climate change may have serious implications for earthworm communities and for the functions they provide.Copyright © 2019 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. No claim to original U.S. Government Works.
Cloning, analysis and functional annotation of expressed sequence tags from the earthworm Eisenia fetida
Eisenia fetida and Eisenia andrei delimitation by automated barcode gap discovery and neighbor-joining analyses: A review
The complete mitochondrial genome sequences of Japanese earthworms Metaphire hilgendorfi and Amynthas yunoshimensis (Clitellata: Megascolecidae)
Genome and single-cell RNA-sequencing of the earthworm Eisenia andrei identifies cellular mechanisms underlying regeneration
DOI:10.1038/s41467-020-16454-8 URL [本文引用: 4]
Variation in nuclear genome size within the Eisenia nordenskioldi complex (Lumbricidae, Annelida)
DOI:10.18699/VJ21.073 URL [本文引用: 1]
Phylogeny of the Eisenia nordenskioldi complex based on mitochondrial genomes
DOI:10.1016/j.ejsobi.2019.103137 URL [本文引用: 1]
Mitochondrial DNA variation in Eisenia n. nordenskioldi (Lumbricidae) in Europe and Southern Urals
There are many peregrine European earthworm species that are found in Siberia. In contrast, it is generally considered that the only Siberian species, E. n. nordenskioldi, was capable to disperse in the reverse direction, from Siberia into Europe. We studied genetic diversity of E. n. nordenskioldi in Southern Urals and Eastern Europe using the mitochondrial cox1 gene. We found that E. n. nordenskioldi from that region represents a new genetic lineage distinct from the previously known populations of this species from Siberia. Molecular clock estimates suggest that this newly found lineage separated from the rest of the species in Lower Pleistocene. Within the studied sample, we detected two geographically restricted groups, which also diverged long before the Holocene, one found in the East European Plain and the other restricted to the Urals. Those two groups were found in sympatry in only one population. Therefore, our results do not support the traditional viewpoint, suggesting that E. n. nordenskioldi is definitely not a recent invader in Europe.
The complete mitochondrial genome of Aporrectodea rosea (Annelida: Lumbricidae)
DOI:10.1080/23802359.2019.1610091 URL
A brief review and evaluation of earthworm biomarkers in soil pollution assessment
DOI:10.1007/s11356-017-8784-0 URL [本文引用: 1]
Earthworms: Charles Darwin’s ‘unheralded soldiers of mankind’: Protective & productive for man & environment
DOI:10.4236/jep.2010.13030 URL [本文引用: 1]
Important issues in ecotoxicological investigations using earthworms
DOI:10.1007/398_2016_4
PMID:27161559
[本文引用: 1]
The importance and beneficial effects of earthworms on soil structure and quality is well-established. In addition, earthworms have proved to be important model organisms for investigation of pollutant effects on soil ecosystems. In ecotoxicological investigations effects of various pollutants on earthworms were assessed. But some important issues regarding the effects of pollutants on earthworms still need to be comprehensively addressed. In this review several issues relevant to soil ecotoxicological investigations using earthworms are emphasized and guidelines that should be adopted in ecotoxicological investigations using earthworms are given. The inclusion of these guidelines in ecotoxicological studies will contribute to the better quantification of impacts of pollutants and will allow more accurate prediction of the real field effects of pollutants to earthworms.
Complete mitochondrial genome of the Burmese giant earthworm
Amynthas corticis genome reveals molecular mechanisms behind global distribution
DOI:10.1038/s42003-021-01659-4 URL [本文引用: 3]
Unearthing the genetic divergence and gene flow of the earthworm Amynthas_YN 2017 sp. (Oligochaeta: Megascolecidae) populations based on restriction site-associated DNA sequencing
DOI:10.1016/j.ejsobi.2020.103210 URL [本文引用: 3]
Complete mitochondrial genome of four pheretimoid earthworms (Clitellata: Oligochaeta) and their phylogenetic reconstruction
DOI:10.1016/j.gene.2015.08.020
PMID:26291739
[本文引用: 1]
Among oligochaetes, the Pheretima complex within the Megascolecidae is a major earthworm group. Recently, however, the systematics of the Pheretima complex based on morphology are challenged by molecular studies. Since little comparative analysis of earthworm complete mitochondrial genomes has been reported yet, we sequenced mitogenomes of four pheretimoid earthworm species to explore their phylogenetic relationships. The general earthworm genomic features are also found in four earthworms: all genes transcribed from the same strand, the same initiation codon ATG for each PCGs, and conserved structures of RNA genes. Interestingly we find an extra potential tRNA-leucine (CUN) in Amynthas longisiphonus. The earthworm mitochondrial ATP8 exhibits the highest evolutionary rate, while the gene CO1 evolves slowest. Phylogenetic analysis based on protein-coding genes (PCGs) strongly supports the monophyly of the Clitellata, Hirudinea, Oligochaeta, Megascolecidae and Pheretima complex. Our analysis, however, reveals non-monophyly within the genara Amynthas and Metaphire. Thus the generic divisions based on morphology in the Pheretima complex should be reconsidered.Copyright © 2015 Elsevier B.V. All rights reserved.
Complete mitochondrial genome of a pheretimoid earthworm Metaphire vulgaris (Oligochaeta: Megascolecidae)
Complete mitochondrial genome of an Amynthas earthworm
Fifteen new earthworm mitogenomes shed new light on phylogeny within the Pheretima complex
DOI:10.1038/srep20096 URL [本文引用: 1]
The complete mitochondrial genome of Lumbricus rubellus (Oligochaeta, Lumbricidae) and its phylogenetic analysis
Earthworm Drawida (Moniligastridae) molecular phylogeny and diversity in Far East Russia and Northeast China
DOI:10.1080/24750263.2020.1741705 URL [本文引用: 1]
Patterns of genetic variation in the Eisenia nordenskioldi complex (Oligochaeta: Lumbricidae) along an elevation gradient in Northern China
DOI:10.3390/d14010035 URL [本文引用: 2]
Characterization of 15 earthworm mitogenomes from Northeast China and its phylogenetic implication (Oligochaeta: Lumbricidae, Moniligastridae)
DOI:10.3390/d14090714 URL [本文引用: 1]
Timing and scope of genomic expansion within Annelida: Evidence from homeoboxes in the genome of the earthworm Eisenia fetida
DOI:10.1093/gbe/evv243 URL [本文引用: 4]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}