


1 引言
DNA条形码是指用于鉴定物种的一个或多个标准化短基因片段(Hebert et al, 2003)。标准化DNA条形码参考数据库的构建需要遵循几个特征, 包括: (1)序列长度控制在目前测序技术可读取的长度范围内; (2)种间序列差异一般应大于种内差异; (3)具有高度保守的侧翼序列以便于扩增引物的设计, 并保证其能覆盖足够多的代表物种; (4)最重要的是其应存在于绝大部分人们感兴趣的物种中(Kress & Erickson, 2012)。目前, 有多个符合这些标准的基因片段被广泛接受并作为DNA条形码应用。例如, 用于动物的有线粒体细胞色素氧化酶亚基(cytochrome c oxidase subunit 1, COI)的一段650 bp的序列(Hebert et al, 2003), 用于植物的有质体核酮糖1,5-二磷酸羧化酶基因(ribulose 1,5-bisphosphate carboxylase gene, rbcL)和成熟酶K基因(maurase K, matK) (Hollingsworth et al, 2009)以及用于真菌的转录间隔区(internal transcribed spacer, ITS) (Nilsson et al, 2009; Schoch et al, 2012) (表1)。
表1 广泛用于DNA条形码技术的标记基因
Table 1
| 标记基因 Marker gene | 目标物种 Targeted group | 数据库 Database |
|---|---|---|
| 16S | 细菌和古细菌 Bacteria and archea (Sogin et al, 2006) | 核糖体数据库项目 Ribosomal Database Project (RDP, Cole et al, 2008); Greengenes (DeSantis et al, 2006); SILVA (Pruesse et al, 2007) |
| ITS | 真菌(Schoch et al, 2012)、植物(Group et al, 2011)、原生生物(Pawlowski et al, 2012) Fungi (Schoch et al, 2012); plant (Group et al, 2011); protist (Pawlowski et al, 2012) | UNITE (Kõljalg et al, 2005); GenBank (Benson et al, 2012) |
| 18S | 原生生物 Protist (Pawlowski et al, 2012) | SILVA (Pruesse et al, 2007) |
| matK + rbcL | 植物 Plant (Hollingsworth et al, 2009) | 生命条形码数据库 Barcode of Life Data Systems (BOLD, Ratnasingham & Hebert, 2007); GenBank (Benson et al, 2012) |
| COI | 动物群(Hebert et al, 2003)、原生生物(Pawlowski et al, 2012) Fauna (Hebert et al, 2003) and protist (Pawlowski et al, 2012) | 核糖体数据库项目 Ribosomal Database Project (RDP, Cole et al, 2008) |
在Hebert等(2003)首次提出DNA条形码的概念后, DNA条形码技术在条形码数量及其应用方面都呈指数增长, 例如作为生物多样性保护的工具用于: 受损标本的物种鉴定(Armstrong & Ball, 2005)、通过肠道内容物和粪便分析食性(Kunz & Whitaker Jr, 1983; Bohmann et al, 2014)、基于环境DNA (environmental DNA, eDNA) (Baird & Hajibabaei, 2012; Taberlet et al, 2012)和生物混合样品或无脊椎动物源DNA (invertebrate-derived DNA, iDNA)样品(Yu et al, 2012; Bohmann et al, 2013; Liu et al, 2013)进行生物多样性评估。这些应用通常需要依赖于高通量测序(HTS)技术, 并被称为宏基因条形码技术(metabarcoding) (Taberlet et al, 2012)。宏基因条形码技术或宏基因组学的方法最初主要应用于微生物学领域, 通过从各种环境样品中提取的DNA来分析表征微生物群落(Caporaso et al, 2011)。过去十多年的研究表明, 此种方法同样可以应用于动物和植物群落(后文中将其称为大生物群落, macrobial community) (Hajibabaei et al, 2016; Deiner et al, 2017)。考虑到微生物宏基因组学研究已经非常成熟, 本文将重点总结和讨论与大生物群落相关的宏基因条形码的技术进展, 主要包括构建DNA条形码参考数据库和DNA序列聚类的一些技术和方法。
2 构建DNA条形码参考数据库
通过过去十多年中全球范围内科学家的通力合作, DNA条形码参考序列数据库, 例如BOLD (Ratnasingham & Hebert, 2007) , 已经初具规模。然而, 目前参考数据库存在的一个典型问题是其数据在地理空间和物种覆盖度方面均存在很大程度上的不平衡, 这主要是由于全球各地在条形码研究方面投入差异所致, 尤其是在物种多样性热点地区, 科研投入尤其是分子生物学相关方面的投入不足, 制约了这些地区物种信息的数字化。尽管HTS平台的单碱基的测序成本显著下降, 但由于测序长度相对较短, 并不适用于对长扩增子测序(例如, COI基因条形码包括引物序列长~719 bp, 而最新的Miseq系统最长可以完成双端300 bp的测序, 仍然无法测通标准条形码序列), 使得Sanger测序仍然是目前获取DNA条形码序列(表1)的主流技术。
不可否认, 随着集中式和工业化的实验室流程的普及, 用于标准DNA条形码技术的分析成本自2000年以来已显著降低(Hebert et al, 2016)。Meier等(2016)通过咨询条形码研究中心, 如安大略生物多样性研究所(Biodiversity Institute of Ontario, BIO)和加拿大DNA条形码中心(Canadian Centre for DNA Barcoding, CCDB), 真实地估算了分析成本。结果表明, 对于具有高质量DNA的样品, 如果不包含额外的服务(例如重新索要收据、二次抽样、返回剩余的组织或DNA等), CCDB的每个样品的商业成本约为20美元。如果序列和标本图片共享给国际生命条形码项目(International Barcode of Life, iBOL), 费用将降至10美元左右。由于Sanger测序技术即将接近其通量和相关化学成本的极限, 因此基于此的条形码测序成本不太可能进一步大幅降低。据估计, 在全球范围内对物种进行条形码登记(Hebert et al, 2016)需要测序1亿个样本。这意味着仅仅构建全球条形码参考序列的预算就需要约10亿美元。参考数据库的不足导致基于HTS的宏基因条形码研究的结论常常只能限于使用分类操作单元(operational taxonomic units, OTUs), 无法确定其具体的物种信息(Linnaean species name), 因而不能将现有的生物学和生态学的知识与用此方法得出的多样性分布规律结合起来。
科学家一直在尝试使用HTS平台以更低的成本和人工投入获取DNA条形码参考序列。为了解决读长的限制, 不同的学者采用了不同的方法, 包括: 获取标准条形码序列的局部序列; 用多轮PCR的方法扩增全长条形码的不同区域; 又或者利用三代单分子高通量测序技术, 如Pacific Biosciences的长读长单分子实时(Single Molecular Real Time, SMRT)测序系统; 通过生物信息学算法来拼装填补由于读长限制而产生的序列中部的缺口(gap)。例如, 早期的研究分别对不同物种单独进行PCR后再混合, 通过Roche 454平台获取物种条形码序列(Shokralla et al, 2014), 但是由于测序通量的限制, 化学试剂成本高, 454平台被迫退出市场, 而且此方法同Meier等(2016)的方法(基于Illumina平台)一样无法获取标准全长的条形码序列。研究人员还试图应用两轮PCR扩增, 每一轮分别扩增全长条形码的部分序列, 随后通过简单拼接获取全长序列(Shokralla et al, 2015; Cruaud et al, 2017)。另外, 单分子测序平台SMRT技术可以获取环形一致序列(circular consensus sequences, CCSs), 对一个分子多次测序, 可以有效校正该平台固有的高测序错误率(10%-16%) (Eid et al, 2008)。因此, 有研究测试了将SMRT技术应用于条形码数据库构建的可行性(Liu et al, 2017; Hebert et al, 2018), 研究测试的混合样品中, 最复杂的样品有来自多达10,000个不同样本的DNA中的COI扩增子。结果表明随着其测序成本的进一步下降, SMRT技术将会是构建DNA条形码数据库方向的强有力的方法之一。Liu等(2017)还提出了一套新的解决方案(HIFI-Barcode), 这是一套准确高效的生物信息学替代方案。该方法可以使研究者基于目前最经济高效的Hiseq平台, 以现有成本的1/10获取全长条形码序列, 而且不需要额外的PCR步骤(表2)。与此同时, 该研究组还利用BGISEQ-500平台最新的单端400 bp (SE 400)测序技术开发了一套简单有效的DNA条形码数据库的实验和分析流程(HIFI-SE) (Yang et al, 2018)。此方法利用测序读长的优势, 可以通过简单的两端序列比对连接从而获得全长的DNA条形码序列。
表2 利用高通量测序平台批量获取DNA条形码的方法
Table 2
| 目标序列长度 Targeted region length (bp) | 优势 Advantages | 劣势 Disadvantages | 参考文献 Reference |
|---|---|---|---|
| ~300 | - | 无法处理较长的目标序列; Roche 454平台 Can not work on long fragments; Roche 454 platform | Shokralla et al, 2014 |
| ~180 | 简单, 易操作, 成本低 Straightforward, easy to operate, cost-efficient | 目标序列偏短, 只能用于物种初筛 Short targeted region; can only be used for species pre-clustering | Meier et al, 2016 |
| ~650 | 标准DNA条形码全长 Standard full-length COI | 普适性差; 需要多轮PCR过程 Poor universality; multiple rounds of PCR | Shokralla et al, 2015; Cruaud et al, 2017 |
| ~650 | 易操作, 标准DNA条形码全长 Easy to operate, standard full-length COI | 相对较高的计算资源 Relatively high requirement for computational resources | Liu et al, 2017 |
| ~650 | 易操作, 标准DNA条形码全长 Easy to operate, standard full-length COI | SMRT平台成本高 High cost of SMRT platform | Hebert et al, 2018 |
| ~650 | 易操作, 标准DNA条形码全长 Easy to operate, standard full-length COI | 测序平台暂时不够普及 Not a mass production | Yang et al, 2018 |
基于高通量技术的条形码参考序列构建方法还体现了更高的灵敏度, 进一步降低了操作成本。虽然目前有对Sanger测序峰图自动读取和识别的程序, 但是Sanger测序往往要求对结果峰图文件进行肉眼观察甄别以优化数据质量, 因此很难开展高效的自动化分析流程。而基于HTS的方法就不存在这样的问题, 使得高效自动化成为可能。此外, 这些基于HTS的方法可以检测微量的PCR扩增子, 因而可以获取那些“失败”的PCR扩增子(电泳凝胶上没有明显条带), 进一步提高了总体条形码成功率(Liu et al, 2017)。虽然所有这些方法都需要在引物上添加样品特异的标签序列, 这会导致在最开始订购引物时产生一次性的费用, 然而一次性的引物合成可以进行数以千次的反应, 每次反应中的引物成本可以少到忽略不计。
虽然上述所有的方法目前都只是在动物类群的COI相关基因中通过验证, 但是根据其方法原理, SMRT技术和HIFI-barcode技术可以很容易转移应用于其他类型的标记基因, 如用于植物的rbcL和matK基因, 但是需要进一步的实验证明。总之, 我们相信, 如果这些新方法在分类学中得到迅速而广泛的应用, 将为全球生物DNA条形码的生成记录开辟新纪元, 最终完成全球生物物种条形码信息的数字化。
3 DNA序列聚类
对DNA分子测序时, HTS平台不同于Sanger测序, 它对结合在测序芯片上的几乎所有DNA分子测序, 分别产出单独的序列。因此, 源自于PCR扩增的错误如单碱基替换和嵌合体, 因为在扩增产物中比例较低, 在Sanger测序中不会被测到, 但是这些问题序列在HTS测序过程中将会被测序并输出为有效序列。这个问题在基于扩增子的生物多样性研究中尤为显著, 导致OTU预测数目大大增加, 产生的序列多样性远远高于抽样的生物群落的“真实”丰富度, 进而高估多样性, 产生不可信的生态发现。如何区分有意义的生物学变异(种内和种间变异)与PCR和测序过程中的错误, 是目前相关分析工具包开发的主要问题, 也是最核心的挑战之一。考虑到现有条形码参考数据库的完整性不足(在物种多样性及单一物种的空间覆盖度方面都有很大的偏向性), 大部分的研究分析都是根据序列相似度采用从头聚类的方法, 以便于真实评估所研究样点的物种多样性信息(alpha diversity), 而非通过比较一个预先准备好的数据库进行多样性分析(Quast et al, 2012; Zhang et al, 2013)。本文将简要介绍几种广泛使用的工具以及其背后的运算逻辑, 以帮助读者更好地理解如何将大规模HTS序列聚类成有生态学意义的OUT (图1)。由于早期很多为Roche 454焦磷酸测序开发的软件已经不再被广泛使用, 将不在本文中进行讨论。
图1
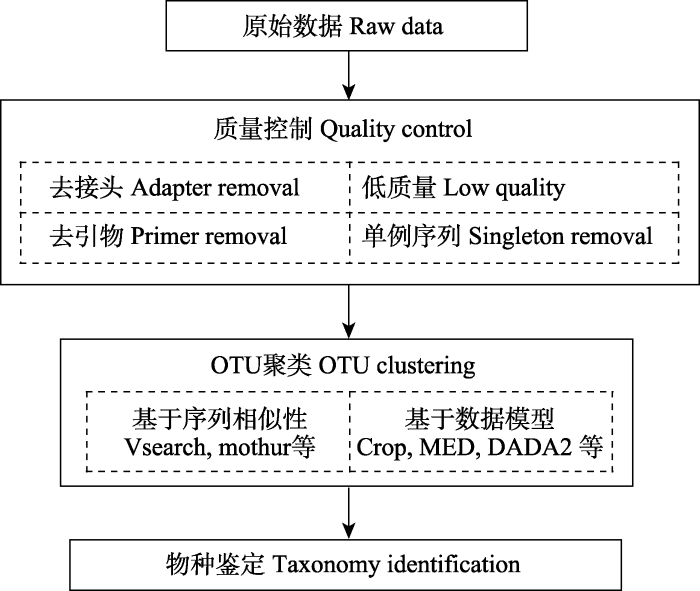
首先, 在进行聚类分析之前, 通过Illumina HiSeq或Miseq测序产生的原始序列需要进行质量过滤, 主要包括去除建库或测序过程中产生的错误序列, 如: (1)含有任何测序接头的序列。在文库制备过程中, 接头序列会添加到插入片段两端。如果模板长度小于测序长度, 测序序列就会含有接头序列。接头也可能出现在序列中间, 这是由于建库过程中引物结合到模板错误的位置, 或者PCR反应中退火, 延伸不足所致。对于前者, 目前常见的可用工具有AdapterRemoval (Schubert et al, 2016)、 Skewer (Jiang et al, 2014)和SOAPnuke (Chen et al, 2017)等, 其中部分软件还可以去除PCR引物, 如果提供标签序列, 还可以进一步拆分样品。如果是后者, 需要将这样的序列整条去除。大多数测序服务供应商都提供接头报告文件帮助用户去除接头序列。(2)含低质量(Q)碱基的序列。Q值平均值虽然是一个广泛采用的参数, 但是在OTU聚类分析中被证明并不是一个可取的办法(Edgar, 2013)。假设有2个相同长度为150 bp的序列, 其质量值分别为140 × Q35 + 10 × Q2和150 × Q25, 那么它们的平均Q值是一样的, 但是前一条序列的预期错误碱基数为6.4, 而后者为0.5。因此, 对于有低Q值的序列, 正确的做法是删除整条的序列, 或删减其末端低质量的序列。
经过序列的预处理之后, 大多数OTU聚类流程, 如U/VSEARCH (Edgar, 2010; Rognes et al, 2016)、 DADA2 (Callahan et al, 2016)、UPARSE (Edgar, 2013)等, 都会进行去除单例序列(singletons)的过程: 首先将相同序列合并为一条代表序列并记录其丰度, 随后去除单例序列(丰度为1的序列)。单例序列被普遍认为是错误序列。此外, 这一步降噪处理还会大大降低序列数量, 从而减轻后续分析的计算负荷。一些软件还包含额外的处理步骤, 如将序列删剪为长度一致的序列(Edgar, 2013)或进行氨基酸翻译检查(Liu et al, 2013)。然而这些额外的降噪处理只适合某些特定的基因, 在使用时需要谨慎, 确认其使用范围。另外, 尽管不同的研究在嵌合体分析中, 有的分析流程倾向于在去除单例序列之后马上进行, 有的倾向于在OTU聚类之后进行, 但是其嵌合体鉴定的原理基本相似, 它们都试图找出双源嵌合体(bimera, two-parent chimera), 即嵌合体序列头尾两端分别来自于同一混合样品中高丰度的其他序列(Edgar et al, 2011; Callahan et al, 2016)。
现已发表的OTU聚类算法大致可以分为两类: (1)首先计算两两序列相似度, 然后以高丰度序列为根序列, 用一个预先设定的相似度(一般为97%)将这些序列进行分组。(2)用数学模型中心化和表征每个聚类单元。传统聚类方法需要计算比较所有的序列来计算距离矩阵, 已经很难处理现阶段研究的数据量(Matias Rodrigues & von Mering, 2013)。而目前广泛流行的USEARCH (Edgar, 2010), 亦或是开源的VSEARCH (Rognes et al, 2016), 或其特定版本的UPARSE (Edgar, 2013)都是基于快速启发式聚类算法。这个算法比传统方法能更快找到与目标序列相似的一个或几个代表序列, 极大降低了计算复杂度。因此目前主流的很多软件采取了类似的聚类方法, 包括: QIIME (UCLUST) (Caporaso et al, 2010)和mothur (Schloss et al, 2009)。CROP是一个基于高斯混合模型(Gaussian Mixture Model)的聚类方法(Hao et al, 2011)。它将高斯分布的平均值替换为一条中心序列, 以此来代表一个特定的组并利用高斯分布来处理测序错误和种间变异。DADA2 (Callahan et al, 2016)开发了一套基于测序质量值的模型, 用于估测Illumina扩增子测序中的错误。首先, 通过对错配碱基和其测序质量值之间进行加权LOESS局部建模(weighted loess fit), 然后通过对数据模型和观测数据的最佳拟合结果将序列聚类。MED (Minimum Entropy Decomposition) (Eren et al, 2015)采用了最小熵分解的算法, 利用序列间的信息不确定性来迭代分解数据集, 直到每个最终待解释的单元满足最大熵标准。
这些基于模型的方法不需要像前述基于序列相似性的聚类方法那样需要预先设定一个临界值(如97%), 而是通过数据本身特性对序列进行聚类。此外, Frøslev等(2017)提出了一个聚类后处理的方法(LULU), 结合序列相似度和共现(co-occurrence)模式从群落数据中去除错误的OTUs。此方法采用了一个类似于MED的算法, 但是其特点在于OTU聚类之后的数据处理, 将在涵盖多时空样本的研究中发挥重要作用。相比于传统基于序列相似度矩阵的方法, 这些基于数学模型的聚类方法可以降低OTU和α多样性高估的状况, 有望找到更多真实的变异, 同时减少错误序列, 从而获得更具有生态学意义的分类单元。得到OTU的参考序列之后, 大部分研究会进一步探讨其物种分类。本文不过多讨论与OTU物种鉴定相关的主题。简而言之, 物种鉴定可以通过使用BLAST或其他类似比对工具, 将序列比对到已建立的参考数据库, 如BOLD (Ratnasingham & Hebert, 2007)、Genbank (Benson et al, 2012)或其他用户定制的数据库。物种分类可以通过最佳匹配以及其序列相似度确定(Shi et al, 2018), 也可以采用基于系统发育树的方法, 根据序列在系统发育树中的位置来确定其物种分类(Zhang et al, 2013)。这两个方法都需要可靠的数据库。如果不能保证数据库的完整度和正确性, 目标序列和数据库数据比对的不确定性将导致模糊的, 甚至是错误的物种鉴定。
4 总结和展望
我们正处于一个物种灭绝速度超过发现速度的时代, 很多物种在得到描述之前就灭绝了。完善正确地描述生物界所有物种的时空分布才能使我们全面了解生物多样性的现状及其驱动力(人为的、自然的, 或两者兼而有之), 进而才能给我们提供有价值的保护和管理策略, 缓解甚至遏止生物多样性持续降低的趋势。条形码技术旨在协助分类学家进行物种鉴定并加速这一过程, 而不是取代传统分类方法(Hebert et al, 2003)。条形码技术具有广泛的适用性(可以应用于生命之树上的几乎所有物种, 无论个体的大小), 而且所需的专业训练不及传统分类学繁重复杂, 使得其不但成为分类学家的工具, 也扩展繁衍出一系列为生态学家和公众服务的工具, 应用于如受损样本和走私货物物种鉴定, 结合HTS技术的生物多样性评估等。另外, 条形码技术不能也不应该局限于基于扩增子的研究分析, 随着测序成本的降低, 条形码技术也在不断发展。略过PCR步骤(Zhou et al, 2013; Tang et al, 2015)和基于捕获芯片的技术(Liu et al, 2016)有望帮助科学家们更准确地理解生物多样性的组成及其驱动的生态过程。基于eDNA和iDNA的相关研究的顺利开展, 将有助于揭示许多物种多样性的分布规律, 尤其是那些小型的、隐秘的物种(Schnell et al, 2012; Mahon et al, 2013; Turner et al, 2014)。然而需要注意的是, 条形码技术及其相关应用更多的是简化了对生态系统中物种鉴定和多样性评估的方法, 但并不能取代合理的生态学设计, 也不能减少样品采集的工作量。研究人员需要根据自身研究的特点设计样品采集方案, 使其最终的研究具有统计学及生物学的意义, 包括采集足够的样点分布和重复个数, 以及涵盖相应的环境参数, 如气候特征和pH梯度等。
参考文献
DNA barcodes for biosecurity: Invasive species identification
DOI:10.1098/rstb.2005.1713 URL [本文引用: 1]
Biomonitoring 2.0: A new paradigm in ecosystem assessment made possible by next-generation DNA sequencing
DOI:10.1111/j.1365-294X.2012.05519.x URL [本文引用: 1]
GenBank
DOI:10.1093/nar/gks1195 URL [本文引用: 3]
Environmental DNA for wildlife biology and biodiversity monitoring
When bugs reveal biodiversity
DOI:10.1111/mec.2013.22.issue-4 URL [本文引用: 1]
DADA2: High-resolution sample inference from Illumina amplicon data
DOI:10.1038/nmeth.3869 [本文引用: 3]
QIIME allows analysis of high-throughput community sequencing data
DOI:10.1038/nmeth.f.303 [本文引用: 1]
Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proceedings of the National Academy of Sciences,
DOI:10.1073/pnas.1000080107 URL [本文引用: 1]
SOAPnuke: A MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. GigaScience, 7, gix120
The Ribosomal Database Project: Improved alignments and new tools for rRNA analysis
High-throughput sequencing of multiple amplicons for barcoding and integrative taxonomy
DOI:10.1038/srep41948 [本文引用: 1]
Environmental DNA metabarcoding: Transforming how we survey animal and plant communities
DOI:10.1111/mec.2017.26.issue-21 URL [本文引用: 1]
Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB
Search and clustering orders of magnitude faster than BLAST
DOI:10.1093/bioinformatics/btq461 URL [本文引用: 2]
UPARSE: Highly accurate OTU sequences from microbial amplicon reads
DOI:10.1038/nmeth.2604 [本文引用: 4]
UCHIME improves sensitivity and speed of chimera detection
DOI:10.1093/bioinformatics/btr381 URL [本文引用: 1]
Real-time DNA sequencing from single polymerase molecules
Minimum entropy decomposition: Unsupervised oligotyping for sensitive partitioning of high-throughput marker gene sequences
DOI:10.1038/ismej.2014.195 [本文引用: 1]
Algorithm for post-clustering curation of DNA amplicon data yields reliable biodiversity estimates
DOI:10.1038/s41467-017-01312-x URL [本文引用: 1]
Comparative analysis of a large dataset indicates that internal transcribed spacer (ITS) should be incorporated into the core barcode for seed plants. Proceedings of the National Academy of Sciences,
DOI:10.1073/pnas.1104551108 URL [本文引用: 2]
A new way to contemplate Darwin’s tangled bank: How DNA barcodes are reconnecting biodiversity science and biomonitoring
DOI:10.1098/rstb.2015.0330 URL [本文引用: 1]
Clustering 16S rRNA for OTU prediction: A method of unsupervised Bayesian clustering
DOI:10.1093/bioinformatics/btq725 URL [本文引用: 1]
A Sequel to Sanger: Amplicon sequencing that scales
DOI:10.1186/s12864-018-4611-3 [本文引用: 1]
Biological identifications through DNA barcodes
DOI:10.1098/rspb.2002.2218 URL [本文引用: 6]
From writing to reading the encyclopedia of life
DOI:10.1098/rstb.2015.0321 URL [本文引用: 2]
A DNA barcode for land plants. Proceedings of the National Academy of Sciences,
DOI:10.1073/pnas.0905845106 URL [本文引用: 2]
Skewer: A fast and accurate adapter trimmer for next-generation sequencing paired-end reads
DOI:10.1186/1471-2105-15-182 [本文引用: 1]
UNITE: A database providing web-based methods for the molecular identification of ectomycorrhizal fungi
DOI:10.1111/j.1469-8137.2005.01376.x URL [本文引用: 1]
DNA barcodes: Methods and protocols. In: DNA Barcodes (eds Kress WJ, Erickson DL), pp.
An evaluation of fecal analysis for determining food habits of insectivorous bats
DOI:10.1139/z83-177 URL [本文引用: 1]
SOAPBarcode: Revealing arthropod biodiversity through assembly of Illumina shotgun sequences of PCR amplicons
DOI:10.1111/mee3.2013.4.issue-12 URL [本文引用: 2]
Mitochondrial capture enriches mito-DNA 100 fold, enabling PCR-free mitogenomics biodiversity analysis
DOI:10.1111/men.2016.16.issue-2 URL [本文引用: 1]
Filling reference gaps via assembling DNA barcodes using high-throughput sequencing—Moving toward barcoding the world
Validation of eDNA surveillance sensitivity for detection of Asian carps in controlled and field experiments
DOI:10.1371/journal.pone.0058316 URL [本文引用: 1]
HPC-CLUST: Distributed hierarchical clustering for large sets of nucleotide sequences
$1 DNA barcodes for reconstructing complex phenomes and finding rare species in specimen-rich samples
DOI:10.1111/cla.2016.32.issue-1 URL [本文引用: 2]
The ITS region as a target for characterization of fungal communities using emerging sequencing technologies
DOI:10.1111/fml.2009.296.issue-1 URL [本文引用: 1]
CBOL protist working group: Barcoding eukaryotic richness beyond the animal, plant, and fungal kingdoms
DOI:10.1371/journal.pbio.1001419 URL [本文引用: 5]
SILVA: A comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB
DOI:10.1093/nar/gkm864 URL [本文引用: 2]
The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools
DOI:10.1093/nar/gks1219 URL [本文引用: 1]
(
VSEARCH: A versatile open source tool for metagenomics
DOI:10.7717/peerj.2584 URL [本文引用: 2]
Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities
DOI:10.1128/AEM.01541-09 URL [本文引用: 1]
Screening mammal biodiversity using DNA from leeches
DOI:10.1016/j.cub.2012.02.058 URL [本文引用: 1]
Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for fungi. Proceedings of the National Academy of Sciences,
DOI:10.1073/pnas.1117018109 URL [本文引用: 3]
AdapterRemoval v2: Rapid adapter trimming, identification, and read merging
DOI:10.1186/s13104-016-1900-2 URL [本文引用: 1]
FuzzyID2: A software package for large data set species identification via barcoding and metabarcoding using hidden Markov models and fuzzy set methods
DOI:10.1111/men.2018.18.issue-3 URL [本文引用: 1]
Next-generation DNA barcoding: Using next-generation sequencing to enhance and accelerate DNA barcode capture from single specimens
Massively parallel multiplex DNA sequencing for specimen identification using an Illumina MiSeq platform
DOI:10.1038/srep09687 [本文引用: 1]
Microbial diversity in the deep sea and the underexplored “rare biosphere”. Proceedings of the National Academy of Sciences,
DOI:10.1073/pnas.0605127103 URL [本文引用: 1]
Environmental DNA
DOI:10.1111/j.1365-294X.2012.05542.x URL [本文引用: 2]
High-throughput monitoring of wild bee diversity and abundance via mitogenomics
DOI:10.1111/2041-210X.12416 URL [本文引用: 1]
Improved methods for capture, extraction, and quantitative assay of environmental DNA from Asian bigheaded carp (Hypophthalmichthys spp.)
DOI:10.1371/journal.pone.0114329 URL [本文引用: 1]
Access COI barcode efficiently using high throughput Single End 400 bp sequencing
Biodiversity soup: Metabarcoding of arthropods for rapid biodiversity assessment and biomonitoring
DOI:10.1111/mee3.2012.3.issue-4 URL [本文引用: 1]
A general species delimitation method with applications to phylogenetic placements
DOI:10.1093/bioinformatics/btt499 URL [本文引用: 2]
Ultra-deep sequencing enables high-fidelity recovery of biodiversity for bulk arthropod samples without PCR amplification
DOI:10.1186/2047-217X-2-4 URL [本文引用: 1]

{kind=link}
{kind=link}