
Biodiv Sci ›› 2019, Vol. 27 ›› Issue (5): 534-542. DOI: 10.17520/biods.2018201 cstr: 32101.14.biods.2018201
• Reviews • Previous Articles Next Articles
Xu Yakun1,2,Ma Yue1,2,Hu Xiaoxi1,Wang Jun1,*( )
)
Received:2018-07-30
Accepted:2018-12-25
Online:2019-05-20
Published:2019-05-20
Contact:
Wang Jun
Xu Yakun, Ma Yue, Hu Xiaoxi, Wang Jun. Analysis of prospective microbiology research using third-generation sequencing technology[J]. Biodiv Sci, 2019, 27(5): 534-542.
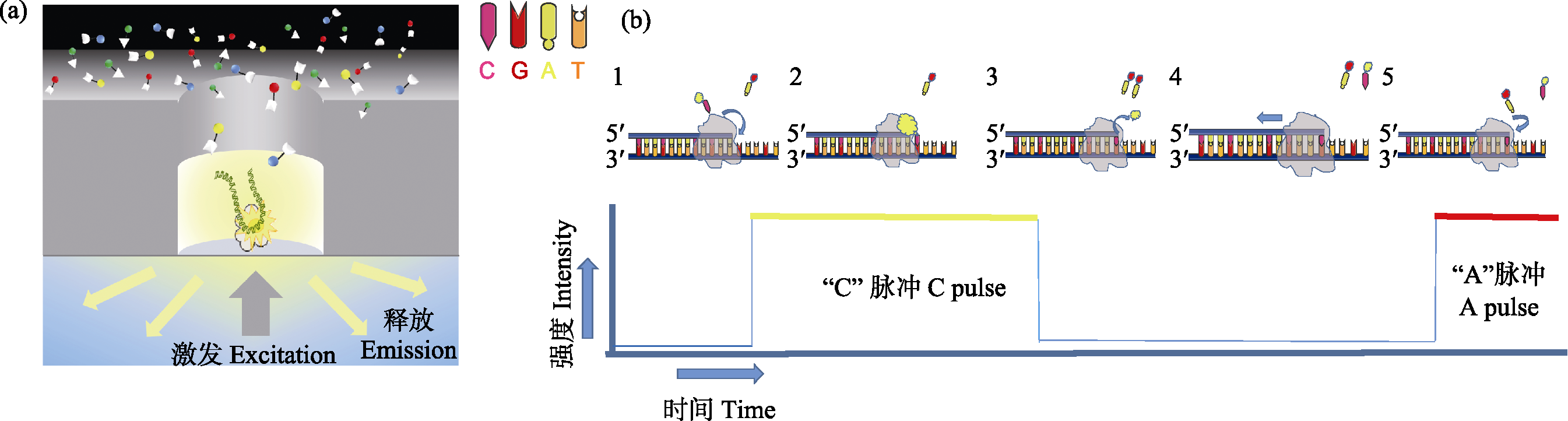
Fig. 1 Schematic diagram of PacBio single molecule real-time sequencing. (a) In the ZMW hole, a single DNA molecule template combined with primers and polymerase is bind to the bottom of ZMW hole. At the beginning of DNA sequencing, the newly added fluorescent labeled dNTP remained at the bottom of ZMW for a long time due to base pairing, and the corresponding fluorescent signals were recorded by confocal microscopy in real time. (b) (1) Fluorescence labeling cytosine deoxynucleotides; (2) Cytosine deoxynucleotides entering DNA chain pairing, emitting fluorescent signals; (3) Fluorescent group is removed by DNA polymerase, fluorescence disappeared; (4) Label new deoxynucleotides; (5) Continue a new round.
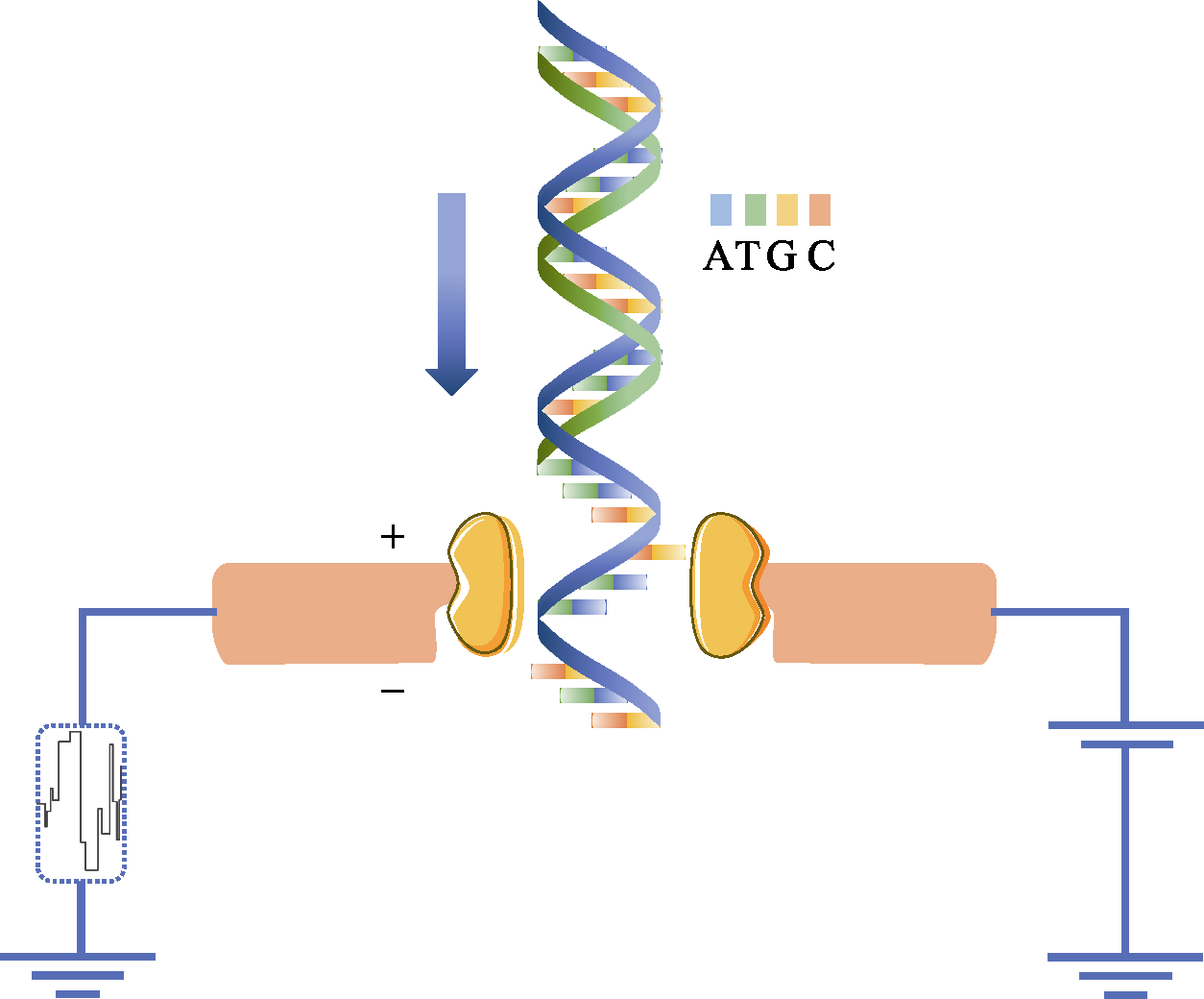
Fig. 2 Nanopore DNA sequencing using electronic signals as detection methods. The diameter of the nanoscale is very small that only a single DNA molecule is allowed to pass through. When a single strand of DNA passes through, it blocks the flow of ions and changes the current intensity across the nanopore. Because the charge properties of the four bases of ATCG are different, the type of base passed is identified according to the change of current.
| 技术平台 Technical platform | 测序原理 Principle of sequencing | 测序读长 Read length | 优点 Advantages | 缺点 Limitations | |
|---|---|---|---|---|---|
| 第一代 The first generation | Sanger | 可中断测序 Chain-terminating sequencing | 600-1,000 bp | 读长长; 准确率高; 能很好地 处理一些重复序列和多聚序列 Long reads; high accuracy; good ability to deal with repetitive and homopolymer regions. | 通量低; 样品制备成本高, 难以做大量的平行测序 Low throughput; high cost of Sanger sample preparation; making massively parallel sequencing prohibitive. |
| 第二代 The second generation | Roche/454 | 焦磷酸测序 Pyrosequencing | 200-400 bp | 在二代测序中读长最长; 高通量 Longest read lengths among the second-generation; high throughput. | 样品制备较难; 难于处理重复和 同种碱基多聚区域 Challenging sample preparation; hard to deal with repetitive/homopo- lymer regions. |
| Illumina | 边合成边测序 Sequencing by synthesis | 2 × 150 bp | 高通量 Very high throughput | 读长短 Short reads | |
| ABI/Solid | 连接测序 Sequencing by ligation | 25-35 bp | 高通量; 成本低 High throughput; low cost. | 测序运行时间长; 读长短, 造成后续 的数据分析困难和基因组拼接困难 Long sequencing runs (days); short reads, resulting in difficulties in subsequence data analysis and genome assembly. | |
| 第三代 The third generation | PacBio SMRT | 边合成边测序/ DNA聚合酶 Sequencing by synthesis/DNA polymerase | ~1,000 bp | 高平均读长; 不需要扩增; 最长单个读长接近100 kb Long average read length; no amplification of sequencing fragments; longest individual reads approach 100 kb. | 错误率高; 依赖DNA聚合酶的活性 Low accuracy; dependence on DNA polymerase activity. |
| Nanopore | 电信号测序/ 核酸外切酶 Electronic signals sequencing/exonuclease | 最大记载2.2 M Maximum record 2.2 M | 读长超长; 电学测序; 方便携带 Over-long read; electronic sequencing; portable. | 错误率高 High sequencing error |
Table 1 Comparison of three generation sequencing technologies
| 技术平台 Technical platform | 测序原理 Principle of sequencing | 测序读长 Read length | 优点 Advantages | 缺点 Limitations | |
|---|---|---|---|---|---|
| 第一代 The first generation | Sanger | 可中断测序 Chain-terminating sequencing | 600-1,000 bp | 读长长; 准确率高; 能很好地 处理一些重复序列和多聚序列 Long reads; high accuracy; good ability to deal with repetitive and homopolymer regions. | 通量低; 样品制备成本高, 难以做大量的平行测序 Low throughput; high cost of Sanger sample preparation; making massively parallel sequencing prohibitive. |
| 第二代 The second generation | Roche/454 | 焦磷酸测序 Pyrosequencing | 200-400 bp | 在二代测序中读长最长; 高通量 Longest read lengths among the second-generation; high throughput. | 样品制备较难; 难于处理重复和 同种碱基多聚区域 Challenging sample preparation; hard to deal with repetitive/homopo- lymer regions. |
| Illumina | 边合成边测序 Sequencing by synthesis | 2 × 150 bp | 高通量 Very high throughput | 读长短 Short reads | |
| ABI/Solid | 连接测序 Sequencing by ligation | 25-35 bp | 高通量; 成本低 High throughput; low cost. | 测序运行时间长; 读长短, 造成后续 的数据分析困难和基因组拼接困难 Long sequencing runs (days); short reads, resulting in difficulties in subsequence data analysis and genome assembly. | |
| 第三代 The third generation | PacBio SMRT | 边合成边测序/ DNA聚合酶 Sequencing by synthesis/DNA polymerase | ~1,000 bp | 高平均读长; 不需要扩增; 最长单个读长接近100 kb Long average read length; no amplification of sequencing fragments; longest individual reads approach 100 kb. | 错误率高; 依赖DNA聚合酶的活性 Low accuracy; dependence on DNA polymerase activity. |
| Nanopore | 电信号测序/ 核酸外切酶 Electronic signals sequencing/exonuclease | 最大记载2.2 M Maximum record 2.2 M | 读长超长; 电学测序; 方便携带 Over-long read; electronic sequencing; portable. | 错误率高 High sequencing error |
| [1] |
Alori ET, Babalola OO ( 2018) Microbial inoculants for improving crop quality and human health in Africa. Frontiers in Microbiology, 9, 2213.
DOI URL |
| [2] |
Benitez-Paez A, Portune KJ, Sanz Y ( 2016) Species-level resolution of 16S rRNA gene amplicons sequenced through the MinION TM portable nanopore sequencer . GigaScience, 5, 4.
DOI URL |
| [3] |
Brown SD, Nagaraju S, Utturkar S, De Tissera S, Segovia S, Mitchell W, Land ML, Dassanayake A, Kopke M ( 2014) Comparison of single-molecule sequencing and hybrid approaches for finishing the genome of Clostridium autoethanogenum and analysis of CRISPR systems in industrial relevant Clostridia. Biotechnology for Biofuels, 7, 40.
DOI URL |
| [4] |
Callaway E ( 2018) Flu virus finally sequenced in its native form. Nature, 556, 420.
DOI |
| [5] |
Chin CS, Alexander DH, Marks P, Klammer AA, Drake J, Heiner C, Clum A, Copeland A, Huddleston J, Eichler EE, Turner SW, Korlach J ( 2013) Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nature Methods, 10, 563-569.
DOI |
| [6] |
Clarke J, Wu HC, Jayasinghe L, Patel A, Reid S, Bayley H ( 2009) Continuous base identification for single-molecule nanopore DNA sequencing. Nature Nanotechnology, 4, 265-270.
DOI |
| [7] | Cusco A, Vines J, D’Andreano S, Riva F, Casellas J, Sánchez A, Francino O ( 2018) Using MinION to characterize dog skin microbiota through full-length 16S rRNA gene sequencing approach. bioRxiv, doi: https://doi.org/10.1101/167015. |
| [8] |
Eid J, Fehr A, Gray J, Luong K, Lyle J, Otto G, Peluso P, Rank D, Baybayan P, Bettman B, Bibillo A, Bjornson K, Chaudhuri B, Christians F, Cicero R, Clark S, Dalal R, Dewinter A, Dixon J, Foquet M, Gaertner A, Hardenbol P, Heiner C, Hester K, Holden D, Kearns G, Kong XX, Kuse R, Lacroix Y, Lin S, Lundquist P, Ma CC, Marks P, Maxham M, Murphy D, Park I, Pham T, Phillips M, Roy J, Sebra R, Shen G, Sorenson J, Tomaney A, Travers K, Trulson M, Vieceli J, Wegener J, Wu D, Yang A, Zaccarin D, Zhao P, Zhong F, Korlach J, Turner S ( 2009) Real-time DNA sequencing from single polymerase molecules. Science, 323, 133-138.
DOI URL |
| [9] | Faino L, Seidl MF, Datema E, van den Berg GC, Janssen A, Wittenberg AH, Thomma BP ( 2015) Single-molecule real-time sequencing combined with optical mapping yields completely finished fungal genome. mBio, 6, e00936-15. |
| [10] |
Fierer N, Breitbart M, Nulton J, Salamon P, Lozupone C, Jones R, Robeson M, Edwards RA, Felts B, Rayhawk S, Knight R, Rohwer F, Jackson RB ( 2007) Metagenomic and small-subunit rRNA analyses reveal the genetic diversity of bacteria, archaea, fungi, and viruses in soil. Applied and Environmental Microbiology, 73, 7059-7066.
DOI URL |
| [11] |
Frank JA, Pan Y, Tooming-Klunderud A, Eijsink VGH, McHardy AC, Nederbragt AJ, Pope PB ( 2016) Improved metagenome assemblies and taxonomic binning using long-read circular consensus sequence data. Scientific Reports, 6, 25373
DOI |
| [12] |
Franzen O, Hu J, Bao X, Itzkowitz SH, Peter I, Bashir A ( 2015) Improved OTU-picking using long-read 16S rRNA gene amplicon sequencing and generic hierarchical clustering. Microbiome, 3, 43.
DOI URL |
| [13] |
Gloor GB, Hummelen R, Macklaim JM, Dickson RJ, Fernandes AD, MacPhee R, Reid G ( 2010) Microbiome profiling by Illumina sequencing of combinatorial sequence-tagged PCR products. PLoS ONE, 5, e15406.
DOI URL |
| [14] |
Greninger AL, Naccache SN, Federman S, Yu GX, Mbala P, Bres V, Stryke D, Bouquet J, Somasekar S, Linnen JM, Dodd R, Mulembakani P, Schneider BS, Muyembe-Tamfum JJ, Stramer SL, Chiu CY ( 2015) Rapid metagenomic identification of viral pathogens in clinical samples by real-time nanopore sequencing analysis. Genome Medicine, 7, 99.
DOI URL |
| [15] |
Huang ZR, Hong JL, Xu JX, Li L, Guo WL, Pan YY, Chen SJ, Bai WD, Rao PF, Ni L, Zhao LN, Liu B, Lv XC ( 2018) Exploring core functional microbiota responsible for the production of volatile flavour during the traditional brewing of Wuyi Hong Qu glutinous rice wine. Food Microbiology, 76, 487-496.
DOI URL |
| [16] |
Hug LA, Baker BJ, Anantharaman K, Brown CT, Probst AJ, Castelle CJ, Butterfield CN, Hernsdorf AW, Amano Y, Ise K, Suzuki Y, Dudek N, Relman DA, Finstad KM, Amundson R, Thomas BC, Banfield JF ( 2016) A new view of the tree of life. Nature Microbiology, 1, 16048.
DOI |
| [17] | Hugenholtz P, Pitulle C, Hershberger KL, Pace NR ( 1998) Novel division level bacterial diversity in a Yellowstone hot spring. Journal of Bacteriology, 180, 366-376. |
| [18] |
Jain M, Koren S, Miga KH ( 2018) Nanopore sequencing and assembly of a human genome with ultra-long reads. Nature Biotechnology, 36, 338-345.
DOI |
| [19] |
Jonasson J, Monstein HJ ( 2002) Classification, identification and subtyping of bacteria based on pyrosequencing and signature matching of 16s rDNA fragments. Apmis, 110, 263-272.
DOI URL |
| [20] |
Liem M, Jansen HJ, Dirks RP, Henkel CV, van Heusden GPH, Lemmers R, Omer T, Shao S, Punt PJ, Spaink HP ( 2017) De novo whole-genome assembly of a wild type yeast isolate using nanopore sequencing. F1000Research, 6, 618.
DOI URL |
| [21] |
Liu H, Hu Z, Zhang Y, Zhang J, Xie H, Liang S ( 2018) Microbial nitrogen removal of ammonia wastewater in poly (butylenes succinate)-based constructed wetland: Effect of dissolved oxygen. Applied Microbiology and Biotechnology, 102, 9389-9398.
DOI |
| [22] | Ludden C, Reuter S, Judge K, Gouliouris T, Blane B, Coll F, Naydenova P, Hunt M, Tracey A, Hopkins KL, Brown NM, Woodford N, Parkhill J, Peacock SJ ( 2017) Sharing of carbapenemase-encoding plasmids between Enterobacteriaceae in UK sewage uncovered by MinION sequencing. Microbial Genomics, 3, 1-12. |
| [23] | Magi A, Semeraro R, Mingrino A, Giusti B, D’Aurizio R ( 2017) Nanopore sequencing data analysis: State of the art, applications and challenges. Briefings in Bioinformatics, 19, 1256-1272. |
| [24] |
Mardis ER ( 2008) Next-generation DNA sequencing methods. Annual Review of Genomics and Human Genetics, 9, 387-402.
DOI URL |
| [25] |
Metzker ML ( 2010) Sequencing technologies—The next generation. Nature Reviews Genetics, 11, 31-46.
DOI |
| [26] |
Niedringhaus TP, Milanova D, Kerby MB, Snyder MP, Barron AE ( 2011) Landscape of next-generation squencing technologies. Analytical Chemistry, 83, 4327-4341.
DOI URL |
| [27] | Payne A, Holmes N, Rakyan V, Loose M ( 2018) Whale watching with BulkVis: A graphical viewer for Oxford Nanopore bulk fast5 files. bioRxiv, doi: https://doi.org/10.1101/ |
| 312256. | |
| [28] |
Quick J, Loman NJ, Duraffour S, Simpson JT, Ettore S, Cowley L, Bore JA, Koundouno R, Dudas G, Mikhail A, Ouedraogo N, Afrough B, Bah A, Baum JHJ, Becker-Ziaja B, Boettcher JP, Cabeza-Cabrerizo M, Camino-Sanchez A, Carter LL, Doerrbecker J, Enkirch T, Garcia-Dorival I, Hetzelt N, Hinzmann J, Holm T, Kafetzopoulou LE, Koropogui M, Kosgey A, Kuisma E, Logue CH, Mazzarelli A, Meisel S, Mertens M, Michel J, Ngabo D, Nitzsche K, Pallasch E, Patrono LV, Portmann J, Repits JG, Rickett NY, Sachse A, Singethan K, Vitoriano I, Emanaberhan RLY, Zekeng EG, Racine T, Bello A, Sall AA, Faye O, Faye O, Magassouba N, Williams CV, Amburgey V, Winona L, Davis E, Gerlach J, Washington F, Monteil V, Jourdain M, Bererd M, Camara A, Somlare H, Camara A, Gerard M, Bado G, Baillet B, Delaune D, Nebie KY, Diarra A, Savane Y, Pallawo RB, Gutierrez GJ, Milhano N, Roger I, Williams CJ, Yattara F, Lewandowski K, Taylor J, Rachwal P, Turner DJ, Pollakis G, Hiscox JA, Matthews DA, O’Shea MK, Johnston AM, Wilson D, Hutley E, Smit E, DiCaro A, Wolfel R, Stoecker K, Fleischmann E, Gabriel M, Weller SA, Koivogui L, Diallo B, Keita S, Rambaut A, Formenty P, Gunther S, Carroll MW ( 2016) Real-time, portable genome sequencing for Ebola surveillance. Nature, 530, 228-232.
DOI |
| [29] |
Sanger F, Nicklen S, Coulson AR ( 1977) DNA sequencing with chain-terminating inhibitors. Proceedings of the National Academy of Sciences, USA, 74, 5463-5467.
DOI URL |
| [30] |
Schloss PD, Handelsman J ( 2005) Metagenomics for studying unculturable microorganisms: Cutting the Gordian knot. Genome Biology, 6, 229.
DOI URL |
| [31] |
Schmid M, Frei D, Patrignani A, Schlapbach R, Frey JE, Remus-Emsermann MNP, Ahrens CH ( 2018) Pushing the limits of de novo genome assembly for complex prokaryotic genomes harboring very long, near identical repeats. Nucleic Acids Research, 46, 8953-8965.
DOI URL |
| [32] |
Singer E, Bushnell B, Coleman-Derr D, Bowman B, Bowers RM, Levy A, Gies EA, Cheng JF, Copeland A, Klenk HP, Hallam SJ, Hugenholtz P, Tringe SG, Woyke T ( 2016) High-resolution phylogenetic microbial community profiling. ISME Journal, 10, 2020-2032.
DOI |
| [33] | Smith AM, Jain M, Mulroney L, Garalde DR, Akeson M ( 2017) Reading canonical and modified nucleotides in 16S ribosomal RNA using nanopore direct RNA sequencing. bioRxiv, doi: https://doi.org/10.1101/132274. |
| [34] |
Sogin ML, Morrison HG, Huber JA, Mark Welch D, Huse SM, Neal PR, Arrieta JM, Herndl GJ ( 2006) Microbial diversity in the deep sea and the underexplored “rare biosphere”. Proceedings of the National Academy of Sciences, USA, 103, 12115-12120.
DOI URL |
| [35] |
Sunagawa S, Coelho LP, Chaffron S, Kultima JR, Labadie K, Salazar G, Djahanschiri B, Zeller G, Mende DR, Alberti A, Cornejo-Castillo FM, Costea PI, Cruaud C, d’Ovidio F, Engelen S, Ferrera I, Gasol JM, Guidi L, Hildebrand F, Kokoszka F, Lepoivre C, Lima-Mendez G, Poulain J, Poulos BT, Royo-Llonch M, Sarmento H, Vieira-Silva S, Dimier C, Picheral M, Searson S, Kandels-Lewis S, Tara Oceans C, Bowler C, de Vargas C, Gorsky G, Grimsley N, Hingamp P, Iudicone D, Jaillon O, Not F, Ogata H, Pesant S, Speich S, Stemmann L, Sullivan MB, Weissenbach J, Wincker P, Karsenti E, Raes J, Acinas SG, Bork P ( 2015) Structure and function of the global ocean microbiome. Science, 348, 1261359.
DOI URL |
| [36] |
Theuns S, Vanmechelen B, Bernaert Q, Deboutte W, Vandenhole M, Beller L, Matthijnssens J, Maes P, Nauwynck HJ ( 2018) Nanopore sequencing as a revolutionary diagnostic tool for porcine viral enteric disease complexes identifies porcine kobuvirus as an important enteric virus. Scientific Reports, 8, 9830.
DOI URL |
| [37] | Tsai YC, Conlan S, Deming C, Segre JA, Kong HH, Korlach J, Oh J, Progra NCS ( 2016) Resolving the complexity of human skin metagenomes using single-molecule sequencing. mBio, 7, e01748-15. |
| [38] | Wick RR, Judd LM, Gorrie CL, Holt KE ( 2017) Completing bacterial genome assemblies with multiplex MinION sequencing. Microbial Genomics, 3, e000132. |
| [39] | Woese CR ( 1987) Bacterial evolution. Microbiological Reviews, 51, 221-271. |
| [40] |
Woese CR, Fox GE ( 1977) Phylogenetic structure of the prokaryotic domain: The primary kingdoms. Proceedings of the National Academy of Sciences, USA, 74, 5088-5090.
DOI URL |
| [41] |
Yang CY, Tarng DC ( 2018) Diet, gut microbiome and indoxyl sulphate in chronic kidney disease patients. Nephrology, 23, 16-20.
DOI URL |
| [42] |
Youssef NH, Couger MB, Struchtemeyer CG, Liggenstoffer AS, Prade RA, Najar FZ, Atiyeh HK, Wilkins MR, Elshahed MS ( 2013) The genome of the anaerobic fungus Orpinomyces sp. strain C1A reveals the unique evolutionary history of a remarkable plant biomass degrader. Applied and Environmental Microbiology, 79, 4620-4634.
DOI URL |
| [43] |
Zhao L, Song Y, Li L, Gan N, Brand JJ, Song L ( 2018) The highly heterogeneous methylated genomes and diverse restriction-modification systems of bloom-forming Microcystis. Harmful Algae, 75, 87-93.
DOI URL |
| Viewed | ||||||
|
Full text |
|
|||||
|
Abstract |
|
|||||
Copyright © 2022 Biodiversity Science
Editorial Office of Biodiversity Science, 20 Nanxincun, Xiangshan, Beijing 100093, China
Tel: 010-62836137, 62836665 E-mail: biodiversity@ibcas.ac.cn