


生物多样性是人类生存和发展的物质基础, 但随着全球气候变化和人类活动的加剧, 自然界中野生动植物多样性急剧降低(Bongaarts, 2019; Ceballos et al, 2020), 很多植物已处于濒临灭绝的边缘(Meyer et al, 2022)。在过去的40多年里, 遗传学已成为研究濒危植物的重要工具。通过对个体和种群遗传变异的分析, 遗传学对保护生物学的多个领域包括遗传多样性量化、物种和亲缘关系的鉴定、有效种群大小的评估、种群亚结构的确定等提供了深刻的认识, 为指导和实施管理策略、缓解濒危风险提供了重要依据(李昂和葛颂, 2001; Primmer, 2009; Fuentes-Pardo & Ruzzante, 2017)。传统上, 保护遗传学研究主要依赖于少数的分子标记如同工酶、微卫星标记或者是细胞器基因组测序(线粒体基因组和叶绿体基因组), 由于使用的大多数分子标记被锚定在基因组上少数的中性区域, 在研究保护生物学问题时有一定的局限性(Avise, 2010; Allendorf, 2017)。
近十几年来, 高通量测序(high-throughput sequencing)技术的发展彻底改变了评估遗传变异的方式(Goodwin et al, 2016; Fuentes-Pardo & Ruzzante, 2017)。这些技术允许在短时间内以可承受的成本对数千到数百万个基因座进行大规模测序, 得到数以万计的高密度分子标记。基因组学技术与保护遗传学理论思想的融合促进了保护基因组学(conservation genomics)的诞生(Allendorf et al, 2010)。与保护遗传学相比, 保护基因组学不仅可以在传统的物种分类地位和亲缘关系的鉴定、遗传多样性、种群遗传结构等方面提供更全面、更准确可靠的研究结果, 还可以对物种起源、分化和种群大小随时间演化的历史进程、种群局部适应的分子机理和近交状况及近交衰退的遗传基础等方面提供更加深入的研究(Allendorf et al, 2010; Fuentes-Pardo & Ruzzante, 2017; Hohenlohe et al, 2021; 魏辅文等, 2021)。
在保护基因组学采用的众多组学方法中, 全基因组重测序(whole genome resequencing, WGR)是目前具有最高分辨力的基因组学方法(Fuentes-Pardo & Ruzzante, 2017)。其优点明显、应用前景广阔, 发展迅速(刘山林等, 2022)。近年来, 有一些综述文章很好地总结了保护基因组学研究取得的一些进展。但是, 这些综述文章更多地聚焦于动物(Fuentes-Pardo & Ruzzante, 2017; 魏辅文等, 2021), 且取得的进展主要来自利用基因组方法中的简化基因组测序方法(reduced-representation genome sequencing, RRGS) (Primmer, 2009; Hohenlohe et al, 2021; 郝宇波等, 2022)。在此, 我们聚焦于利用全基因组重测序方法在濒危植物保护研究方面取得的一些进展。我们首先关注全基因组重测序方法如何提供更多的分子标记来更全面、精准地研究传统的保护遗传学问题如遗传多样性、系统发育关系及群体遗传结构等, 然后进一步关注全基因组重测序方法如何深入地揭示濒危植物有效种群大小随时间的演化历史, 种群适应性演化的分子基础和近交衰退的遗传基础。最后, 本文对重测序方法应用于濒危植物保护研究中存在的问题及未来的发展趋势提出进一步的思考和建议。
1 保护基因组学方法和策略
传统的保护遗传学研究, 依赖于包括等位酶和微卫星基因分型或线粒体DNA测序在内的技术, 以提供关于自然种群的丰富知识。然而, 这些技术只能提供非常有限的遗传标记数据。21世纪以来, 测序技术特别是二代及三代测序技术的飞速发展, 促进了保护基因组学的诞生。目前, 被广泛应用于保护基因组学的方法主要可以分为两大类: 简化基因组测序和全基因组测序(Fuentes-Pardo & Ruzzante, 2017)。
1.1 简化基因组测序
简化基因组测序即对部分基因组进行序列测定, 它极大地降低了基因组的复杂度, 进而降低了测序成本和计算的负担, 此外与全基因组测序相比, 还具有性价比高、稳定性好、文库的构建程序更简单、实验时间较短、得到SNPs (single nucleotide polymorphism)数量多、不依赖于参考基因组等众多优点, 因此该技术被广泛应用于濒危动植物的保护基因组学研究(Fuentes-Pardo & Ruzzante, 2017; Wang et al, 2019; Bao et al, 2020; Cai et al, 2021)。简化基因组测序可分为: 酶切位点相关DNA测序(restriction-site associated DNA sequence, RAD-seq) (Andrews et al, 2016), 转录组测序(RNA-sequencing, RNA-seq) (祁云霞等, 2011), 全外显子组测序(whole exome sequencing, WES) (Warr et al, 2015)。这3种方法的共同点是它们通常只评估基因组的一小部分。由于基因组的不完全覆盖和一些数据的缺失, 简化基因组数据给后续的种群遗传学分析带来了挑战, 例如在群体系统发育推断方面: 首先, 当存在多态性和测序错误的情况下, 很难快速准确地对来自同一限制性位点进行聚类; 其次, 从头将每个聚类组装成独特的位点最终构建系统发育关系仍然比较复杂; 此外, RAD-seq数据最终获得遗传变异信息的规模以及系统发育的可用性, 均受到所用的限制性酶、所选片段的大小以及不同样品的测序覆盖率等多种因素的影响(Chong et al, 2012; Eaton, 2014; Andrews et al, 2016)。而相较于简化基因组测序方法, 依赖于参考基因组的全基因组重测序方法使检测获得的标记的数量和质量均有显著提高, 它极大地优化获得遗传标记的准确度(Andrews et al, 2016; Jones & Good, 2016)。
1.2 全基因组测序
全基因组测序可分为两类: 全基因组从头测序(de novo whole-genome sequencing)和全基因组重测序(whole genome resequencing) (Fuentes-Pardo & Ruzzante, 2017)。从头测序是指首次组装一个新的基因组序列, 基因组组装的难度和质量取决于基因组的大小和复杂性、计算资源和生物信息学经验。目前, 全基因组从头测序主要采用三代测序技术, 有Pacific Biosciences公司的单分子实时测序(single-molecule real-time sequencing, SMRT)技术、和Hifi (high fidelity reads)技术、Nanopore公司的纳米孔测序技术(Oxford nanopore technologies, ONT) (Schuster, 2008; Clarke et al, 2009), 在测序完成后Hi-C (high-throughput chromosome conformation capture)技术则可以帮助将测序结果组装到染色体水平。全基因组重测序的目的是比较个体和种群的基因组变异。全基因组重测序主要利用二代测序技术Roche 454 GS FLX Titanium、ABI公司的SOLID和Illumina公司的HiSeq 2000等获得大量的短片段(short-reads)并比对到参考基因组上从而获得群体水平的单核苷酸多态性(single nucleotide polymorphism, SNP)数据, 然后在此基础上进行一系列的种群遗传学分析。
全基因组重测序需要高质量的参考基因组来进行读长比对和变异检测。高质量参考基因组的缺乏是全基因组重测序技术用于保护生物学研究的主要限制因素。虽然测序技术的高速发展促进了大量高质量参考基因组的解析, 但受限于成本、技术, 无法对所有物种做到均匀覆盖, 往往具有较高的物种以及地理偏向性(刘山林等, 2022)。在此, 我们归纳整理了记录在《国家重点保护植物名录2021》和IUCN红色名录上受威胁植物的全基因组从头测序情况(图1)。相比于已经测序的其他植物物种, 受威胁植物所占的比例很小。尽管如此, 随着人们保护意识的增强和一些重要项目的启动, 预计将来会有越来越多的濒危植物基因组被解析出来。如地球生物基因组计划(Earth BioGenome Project)提出优先对IUCN红色名录收录的23,000多种濒危物种基因组进行测序(Lewin et al, 2018)。这一项目的实施, 将为濒危物种的保护基因组学研究提供助力。
图1
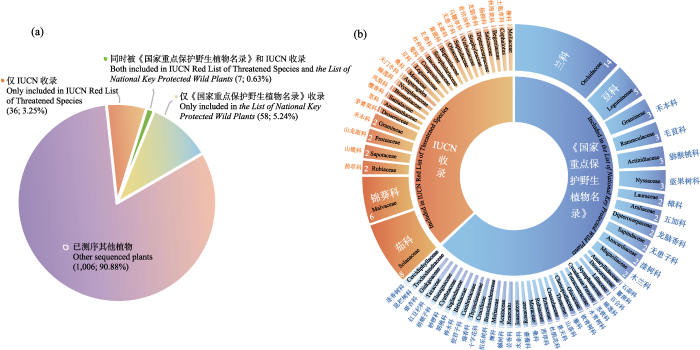
图1
全基因组从头测序的受威胁植物统计。(a)已经全基因组测序的受威胁植物饼图。括号中的数字分别表示已经全基因组测序的受威胁植物物种数量及其所占总测序植物物种数量的比例。(b)《世界自然保护联盟红皮书》和2021年发布的《国家重点保护野生植物名录》收录的受威胁植物所属科统计图。图中的数字表示物种的数量。
Fig. 1
The species statistics with de novo whole-genome sequencing. (a) Comparison of threatened plant species and other plant species with de novo whole-genome sequencing. Values in bracket indicate the number of threatened plant species with de novo whole genome sequencing and its proportion to the total plant species with de novo whole genome sequencing. (b) The distribution of species with de novo whole-genome sequencing included in IUCN Red List of Threatened Species and the List of National Key Protected Wild Plants (2021) in different family. Values indicate the number of species in each family.
2 全基因组重测序在濒危植物保护中的应用
2.1 系统发育关系和种群遗传结构
保护计划的成功实施在很大程度上依赖于对保护目标分类地位的正确识别。全基因组数据记录了一个物种进化过程中的全部历史。通过比较基因组的大部分数据而不是像传统方法那样的少数几个基因, 可以构建更加稳健的系统进化关系, 为近缘物种的识别和隐存种的发现提供了新的解决办法(Fuentes-Pardo & Ruzzante, 2017)。例如, 内蒙古珍稀濒危保护植物互叶醉鱼草(Buddleja alternifolia)主要分布于喜马拉雅、横断山和黄土高原三大区域。其中, 黄土高原地区与其他两个地区在形态上存在显著差异, 而分布于喜马拉雅和横断山的种群在形态上没有区别, 无法判断分布于三个地区的群体是否属于3个物种。Ma等(2021b)首先组装了互叶醉鱼草高质量基因组, 然后获得了分布于三大区域48个居群的样本重测序数据。他们发现三大区域的互叶醉鱼草形成了与地理分布相一致的3个独立的明显分支, 且种群分化系数FST均大于0.5, 他们推测这3个地区的互叶醉鱼草应属于3个不同的种。同样地, 人们利用保护基因组学的方法在濒危动物中也发现了许多类似的例子, 虽然某些濒危动物从形态上无法判断是否属于不同的种, 但是遗传分化已经非常大, 实际上可以定位为不同的物种(谭鑫鑫和李明, 2018)。对这些隐存种的识别对于濒危动植物的保护来说尤其重要。如在互叶醉鱼草的例子中, 本就已经濒危的互叶醉鱼草在基因组上被确认为3个物种, 意味着每个新种比之前预测的数量更少、分布范围更窄, 物种的濒危程度可能更高。在实施保护管理时, 这3个可能的物种也应该分别进行管理。
同一物种的不同种群之间往往存在遗传多样性的分化。保护生物学的目标之一是尽可能地保护易危物种的遗传多样性。种群管理最重要的一步是确定和划定种内保护单位(conservation units, CUs)的边界, 如进化显著单位(evolutionarily significant units, ESUs)。进化显著单元代表了一个物种内种群间的绝大部分遗传多样性(Funk et al, 2012; Forester et al, 2022)。除了ESUs, 还有管理单元(management units, MUs)和适应性单元(adaptive units, AUs)。在基因组框架内, Funk等(2012)建议用所有可能位点的遗传结构分析来鉴定ESUs, 而用中性位点来确定MUs, 用适应性位点来确定AUs。目前, 利用所有的重测序数据进行群体遗传结构分析是种群基因组学的常规分析(Zhao et al, 2019; Liu et al, 2020; Ma et al, 2021a, b), 其结果可以作为濒危植物ESUs划分的依据。Liu等(2020)利用了SMRT和Hi-C技术对水青树(Tetracentron sinense)进行了全基因组从头测序和染色体水平的组装, 并对分布于中国代表性区域的55个水青树个体进行了重测序和群体遗传结构分析(Structure、PCA和NJ建树等分析), 发现水青树具有4种古老的遗传组分, 对应于4个不同的ESUs。Liu等(2022)对分布在中国的13个伯乐树(Bretschneidera sinensis)群体的154个个体进行重测序, 利用去除选择位点后的中性位点来进行结构分析, 将中国的伯乐树群体分为西部群体和东部群体, 对应于两个MUs。
上述研究表明, 基因组重测序数据可以为系统发育关系的重建和种群遗传结构的划分提供强有力的统计支持, 有效地解决使用传统方法无法解决的模糊不清的系统发育关系和种群遗传结构, 很好地确定保护对象和保护单元, 对物种保护具有重要意义。
2.2 基因组多样性的评估
遗传多样性与物种的适应性演化和演化潜能密切相关。因此, 濒危植物的遗传多样性一直都是保护生物学关注的重点。传统的保护遗传学对种群遗传多样性的评估主要基于同工酶、细胞器基因组以及其他的分子标记, 但是这些方法仅能从群体中获得有限的遗传变异资源。采用全基因组重测序方法可以得到一个物种几乎全部的遗传信息, 可以整体评估物种或者种群的遗传多样性即基因组多样性。
近年来, 随着濒危植物基因组不断地被解析, 以及基于重测序的种群基因组数据的积累, 一些濒危植物的基因组多样性被评估。在此, 我们搜索和统计了目前已进行过重测序研究的受威胁植物和无危植物的基因组多样性(表1, 附录1)。相比于一些无危植物, 受威胁植物的遗传多样性相对偏低(图2)。同时, 受威胁植物物种之间的遗传多样性水平存在差异。其中, 斧翅沙芥(Pugionium dolabratum)和水青树具有最高的遗传多样性, 其次遗传多样性较高的为野生荔枝(Litchi chinensis)、茶(Camellia sinensis)和珙桐(Davidia involucrata)。水青树和珙桐为第三纪孑遗植物, 它们较高的遗传多样性可能与其漫长的进化历史有关。较高的遗传多样性说明这些物种具有较高的进化潜能。在这些受威胁植物中,濒危植物芒苞草(Acanthochlamys bracteate)具有最低的遗传多样性。研究也发现, 有些活化石植物如银杏(Ginkgo biloba)尽管形态变异比较少, 但是遗传多样性仍比较高, 显示银杏仍有相当的进化潜能, 且形态变异与遗传变异之间没有相关性(Zhao et al, 2019)。值得注意的是, 有些研究群体取样较少, 代表性不足, 这也可能导致了不同濒危植物检测到的遗传多样性水平的差异。未来濒危植物的保护基因组学研究需要加大取样量从而更全面地评估物种或种群水平的基因组多样性。
表1 受威胁植物核酸多样性
Table 1
| 学名 Scientific name | 濒危等级 Endangered category | 保护等级 Protection level | θπ | 样本量 Sample size | 参考文献 References |
|---|---|---|---|---|---|
| 野生稻 Oryza rufipogon | 极危 CR | Ⅱ级 Class II | 0.003000 | 446 | Huang et al, 2012 |
| 天目铁木 Ostrya rehderiana | 极危 CR | I级 Class I | 0.001660 | 13 | Yang et al, 2018 |
| 野生莲 Nelumbo nucifera | - | II级 Class II | 0.002177 | 7 | Huang et al, 2018 |
| 鹅掌楸 Liriodendron chinense | - | II级 Class II | 0.001280 | 14 | Chen et al, 2019 |
| 银杏 Ginkgo biloba | 极危 CR | I级 Class I | 0.002110 | 545 | Zhao et al, 2019 |
| 水青树 Tetracentron sinense | II级 Class II | 0.012800 | 55 | Liu et al, 2020 | |
| 细叶杨 Populus ilicifolia | 易危 VU | - | 0.000830 | 19 | Chen Z et al, 2020 |
| 珙桐 Davidia involucrate | - | I级 Class I | 0.005850 | 10 | Chen Y et al, 2020 |
| 连香树 Cercidiphyllum japonicum | - | II级 Class II | 0.001100 | 82 | Zhu et al, 2020 |
| 小粒咖啡 Coffea arabica | 濒危 EN | - | 0.003810 | 48 | Huang et al, 2020 |
| 朱红大杜鹃 Rhododendron griersonianum | 极危 CR | - | 0.001940 | 31 | Ma et al, 2021a |
| 胡桃 Juglans regia | 易危 VU | II级 Class II | 0.000500 | 55 | Bernard et al, 2021 |
| 斧翅沙芥 Pugionium dolabratum | - | II级 Class II | 0.013000 | 20 | Hu et al, 2021 |
| 茶 Camellia sinensis | - | II级 Class II | 0.006100 | 120 | Lu et al, 2021 |
| 野生荔枝 Litchi chinensis | - | II级 Class II | 0.008300 | 38 | Hu et al, 2022 |
| 漾濞槭 Acer yangbiense | 濒危 EN | - | 0.003130 | 105 | Ma et al, 2022 |
| 芒苞草 Acanthochlamys bracteata | 易危 VU | - | 0.000460 | 14 | Xu et al, 2022 |
| 野大豆 Glycine soja | - | II级 Class II | 0.001220 | 40 | Wang et al, 2022 |
濒危等级来源于IUCN濒危等级。保护等级来源于2021年发布的《国家重点保护植物名录》。θπ: 群体中任意两条不同序列(个体)的碱基差异数(SNP)的平均值。
Endangered category was retrieved from IUCN endangered level. Protection level was retrieved from the List of National Key Protected Wild Plants in 2021. θπ: Average number of pairwise nucleotide differences per site; CR: Critically endangered, VU: Vulnerable, EN: Endangered.
图2
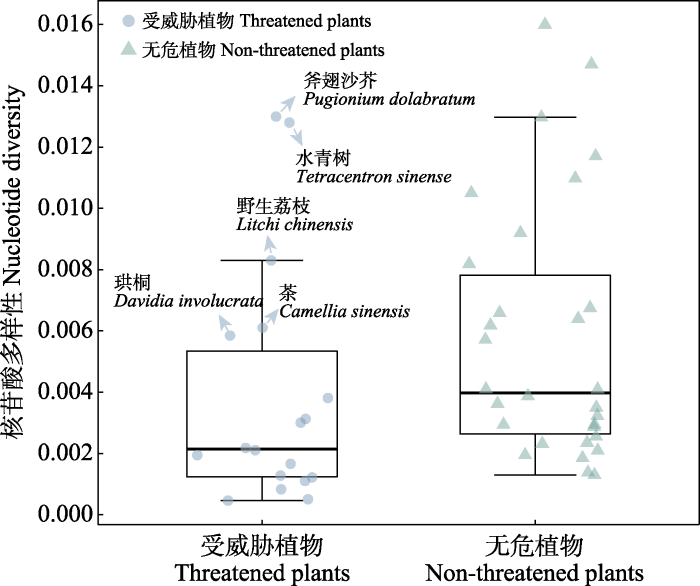
图2
受威胁植物和无危植物的基因组多样性(π)统计。图中只显示遗传多样性最高的5种受威胁植物的名称。
Fig. 2
Genome-wide nucleotide diversity (π). Only species names of five threatened species with the highest genetic diversity are shown.
2.3 种群动态历史
种群动态历史研究物种种群大小和相关参数随着时间变化的情况。濒危植物有效种群动态历史的研究(包括历史有效种群大小的扩张和收缩、瓶颈形成模式、迁移模式等)有助于揭示植物的濒危过程和原因, 对濒危植物的保护具有重要意义。传统的保护遗传学借助线粒体和叶绿体DNA片段或者微卫星遗传标记来推断种群历史动态, 需要的群体样本量大, 且仅能追溯最近一次的种群动态事件(魏辅文等, 2021)。而全基因组重测序数据可以全面重建种群大小在不同时间尺度上变化的历史动态, 为我们了解过去的历史事件对当代有效种群数量以及遗传组成的影响提供了新的见解。
Yang等(2018)对铁木属(Ostrya)的两个近缘种天目铁木(O. rehderiana)和多脉铁木(O. chinensis) 共26个个体进行了重测序, 并对测序数据进行了种群动态历史分析, 发现这两个物种在前期经历了相似的种群动态历史, 发生过两次群体大小骤缩。但是, 末次盛冰期(last glacial maximum)后, 这两个物种遵循了不同的进化路径, 广布种多脉铁木在冰期结束后有效群体大小迅速回升, 而濒危种天目铁木在冰期结束后有效群体大小持续下降, 现已濒临灭绝, 他们推测濒危种天目铁木种群的崩溃可能是由历史气候变化和人为干扰共同造成的。在银杏、珙桐、水青树和伯乐树等物种中, 研究也发现了多种种群动态历史事件, 如种群扩张、种群瓶颈的遗传证据, 推断历史上多次冰期很可能是它们有效群体大小下降的原因(Zhao et al, 2019; Chen Y et al 2020; Liu et al, 2020; Liu et al, 2022)。冰期可造成物种有效群体大小的下降, 而间冰期气温的恢复可能有利于种群的扩张。Chen等(2018)利用PSMC软件重建了鹅掌楸(Liriodendron chinense)和北美鹅掌楸(L. tulipifera)的群体动态历史, 发现在整个第四纪冰期, 北美鹅掌楸的种群数量持续减少, 而鹅掌楸大约在0.4 mya时种群得到恢复并达到峰值, 这两个物种经历的不同群体动态历史很大程度上解释了北美鹅掌楸遗传多样性为何严重丢失而中国鹅掌楸遗传多样性相对较高。鹅掌楸的种群数量恢复时间0.3-0.4 mya与古乡冰期(Guxiang glaciation, 0.3-0.13 mya)和聂聂雄拉冰期(Naynayxungla glaciation, 0.72-0.5 mya)之间的间冰期时间一致, 推测间冰期的温度恢复和冰雪消融作用为东亚避难所内鹅掌楸种群的恢复提供了基础。除了气候因素(如冰期与间冰期), 地质历史事件和人为干扰也是造成种群大小波动的重要原因。Ma等(2021a)基于31个个体的重测序数据对朱红大杜鹃(Rhododen- dron griersonianum)进行了群体动态历史的分析, 发现反复的遗传瓶颈效应是导致朱红大杜鹃相比近缘广布种马缨杜鹃(Rhododendron delavayi)遗传多样性低的重要因素。朱红大杜鹃历史上经历了3次严重遗传瓶颈, 与冰期-间冰期作用、“共和运动”等地质历史事件一致, 最后一次遗传瓶颈之后, 朱红大杜鹃的有效种群大小逐渐恢复, 但是现代的人为干扰可能是造成了该物种目前种群规模较小和分布受限的主要因素。除此之外, 物种在冰期的地理分布可能也影响种群的动态历史。Wang等(2022)对从中国三大农业生态区采集的185种野大豆(Glycine soja)种质进行了全基因组测序, 种群动态历史分析发现野生大豆的有效种群大小自0.6 mya到0.2 mya持续减少后, 所有种群的有效种群大小持续发生不同程度地扩大, 末次冰期并未造成种群大小的减少。Wang等(2022)认为前期种群的持续减少可能是由聂聂雄拉冰期的低温造成的, 而在末次冰期时野生大豆种群非减反增的原因可能是因为此时野生大豆主要分布在温暖的华南地区, 适宜的生长环境促进了其有效种群大小的持续扩张。
总之, 全基因组重测序可以帮助推断有效群体的波动和追踪种群动态历史事件, 并推断过去的地质气候等历史事件对当代有效种群数量以及遗传组成的影响, 如上述研究表明极端的地质气候变化和人类的活动是造成物种有效群体大小急剧减少和遗传多样性降低的重要原因。将种群大小变化与历史环境变化联系起来, 还可以帮助预测未来环境变化对种群分布和遗传多样性的影响。
2.4 选择信号和种群局域适应
植物的适应性变异决定了它们的长期生存能力、种群大小和分布增加的潜能以及灭绝的概率。除了评估适应性表型性状是否有遗传基础外, 基于重测序的保护基因组学还可以确定自然群体中这种变异背后的特定基因, 并且使研究者可以更好地理解适应的过程和潜力。
目前, 在基因组水平上, 通常有两类方法来检测基因组上的选择信号: (1)基于群体间遗传分化指数FST的“离群值”检测方法; (2)基于等位基因频率和环境变量之间相关性的基因型-环境互作关系检测法(Hohenlohe et al, 2021)。前一种方法的理论依据是: 受到适应性选择或正选择的位点明显比基因组上中性进化的其他区域有更高的遗传分化。第二种方法则考虑到不同群体之间存在的环境异质性问题, 旨在将等位基因频率的模式与环境梯度联系起来。基于两类方法的软件有很多, 如BayeScan、Fdist2、“LEA” R package和SAM Matlab等(Beaumont & Nichols, 1996; Vitalis et al, 2001; Joost et al, 2007; Frichot et al, 2013)。此外, 确定自然种群适应性变异的遗传基础的其他方法有全基因组关联研究(genome-wide association studies, GWAS)。基于这些方法, 多个濒危物种中受选择或与地方性适应相关的候选基因被鉴定出来。Zhao等(2019)利用SweeD软件并结合CLR统计在银杏东部和西南群体中分别检测到了910和949个受选择区域, 分别包含了643和504个候选基因, 进一步结合FST分析, 鉴定出25个基因, 这些基因主要参与对昆虫和真菌的抗性以及对失水、低温和高盐等非生物胁迫的响应。环境适应性相关变异位点的鉴定以及生物学过程的富集对于后期的遗传拯救(例如, 引种过程中不同生态区域的环境适应)提供了理论指导(Shear & Werth, 2014; Combosch et al, 2017)。Hu等(2021)发现沙芥(Pugionium cornutum)和斧翅沙芥的基因组中存在42个遗传分化较大的区域, 而对该区域进行分析后鉴定出197个受选择基因, 这些基因参与植物根的发育、叶片形态构成、木质部的分化、种子发育、耐盐碱、耐干旱、氧化应激反应和类黄酮生物合成等等。该研究认为这些受选择的基因在这两个物种形态分化和各自适应当地生态小环境过程中起到了重要作用。Zhu等(2020)通过对连香树(Cercidiphyllum japonicum)日本和中国群体的FST和基因型-环境关联分析等鉴定出的823个与地方性适应可能相关的基因, 它们主要富集在细胞的发育和增殖、生长素代谢途径和胁迫响应等方面。基因型-环境关联方法也被用于检测伯乐树和野生大豆的局域适应。Liu等(2022)利用PCAdapt和BayPass软件对伯乐树群体数据进行基因组扫描筛选出388个受选择的SNP位点, 涉及的基因可能与伯乐树的生长(如PLIM2B_1、JHS1和MES17基因)和胁迫应答(如VTE5、RH1_3和cycl11_1基因)有关。Liu等(2022)认为伯乐树(B. sinensis)中受正选择的基因主要参与胁迫反应与生长相关等生物学过程, 表明该物种仍然具有多种适应潜力支持其持久存在。Wang等(2022)使用了两种基因型-环境关联方法对分布于中国三大农业生态区的185种野大豆种质进行了基因组扫描, 发现了多个参与局域适应的基因, 如开花时间和温度相关基因。在第19染色体上发现了一个经历了正选择的位点, 该位点含有两个相邻MADS-box转录因子, 可能与野大豆能够适应高纬度环境有关。在铁皮石斛(Dendrobium officinale)和古茶等濒危植物中, GWAS则被用来检测受选择的位点。Niu等(2021)对来自13个地区的铁皮石斛及其5个近缘种的38个个体样本进行了重测序并结合GWAS分析, 识别出13个GWAS位点, 其中这些位点包含了4个与株高性状有关的基因, 2个与叶长性状有关的基因, 3个与茎长性状有关的基因和1个与节间长性状有关的基因以及其它的未鉴定功能基因。这些基因中MWL1基因是木质素生物合成的关键基因, 参与次生细胞壁的形成。敲除MWL1基因及其近缘基因MWL2后, 株高显著降低。推测MWL1基因很可能与铁皮石斛的植株产量(茎产量)有关。Lu等(2021)对来自云南和贵州的120棵古茶进行了GWAS分析,发现了4个与叶形相关、两个与株型相关的基因。
综上, 全基因组重测序方法是检测自然选择信号、揭示表型性状的遗传基础和鉴定地方适应的有利武器。它可以促进我们对遗传变异和适应特性内在机制的理解, 有助于我们采取有针对性的保护措施促进濒危植物对快速改变的自然环境的适应。
2.5 其他(近交衰退、有害突变等)
除了能够鉴定出适应环境变化的基因位点外, 全基因组重测序方法还可以揭示小种群适合度降低的遗传基础。小种群由于遗传漂变会导致有害突变积累, 而近交导致纯合有害等位基因的比例增加, 从而降低了个体的适合度。全基因组重测序方法不仅可以用来评估和检测近交事件, 还可以揭示造成群体适合度降低的有害突变位点和与之相关联的基因。
目前, 人们通常基于基因组连续性纯合片段ROHs (runs of homozygosity)计算获得的基因组近交系数FROH (frequency runs of homozygosity, FROH)来评估近交事件(Hohenlohe et al, 2021)。连续性纯合片段, 即染色体上很少或没有杂合核苷酸位点的区域。ROHs的出现通常是由于个体继承了来自父母双方相同的单倍体型, 而这个单倍型又是从过去某个时间点的共同祖先继承而来。短ROH反映了较老的近交事件, 而较长的ROH则反映了近期的近交事件。目前, 也有多种软件可以用来检测群体基因组水平上的有害突变。比如, 基于序列同源性的SIFT预测软件可以帮助分析新出现的非同义变异是否为有害突变(Ng & Henikoff, 2003); 基于序列同源性和蛋白质结构的PolyPhen-2预测软件可以帮助预测群体中有害的错译突变(Adzhubei et al, 2010)。利用这些软件, 人们检测了天目铁木、漾濞槭(Acer yangbiense)和朱红大杜鹃等濒危植物种群的近交衰退和有害突变情况。Yang等(2018)通过对铁木属濒危种天目铁木和广布近缘种多脉铁木的ROH分析, 发现濒危种天目铁木的FROH (0.31-0.45)比广布近缘种多脉铁木(0.07-0.19)要高, 且每个天目铁木个体有几个大于1 Mb的ROHs, 而近缘种多脉铁木最长的ROH不超过0.63 Mb, 推测天目铁木相对于近缘种多脉铁木存在高度近亲交配, 加剧了种群基因组遗传多样性的降低。遗传负荷分析显示天目铁木存在较高的遗传负荷积累, 综合4类(SYN、TOL、DEL、LoF)突变位点于两个铁木树种中的比较情况, 确定天目铁木显著携带更多的纯合有害突变, 这可能是天目铁木濒危的主要原因之一。但天目铁木要比近缘种多脉铁木清除了更多严重有害的隐性突变, 这种清除和逐渐降低的近交衰退有可能一起减缓该物种的灭绝, 并可能促进杂交天目铁木在未来的存活。Yang等(2018)对天目铁木的基因组遗传多样性被侵蚀的模式调查为后续该物种的保护提供了一个很好的例子, 认为未来应该设计人工杂交策略, 以减少由于近亲繁殖和遗传漂移造成的多样性损失, 而不是通过收集近亲繁殖的种子或在濒危树木中进行克隆扦插来增加存活个体的总数。Ma等(2021a)对极度濒危物种朱红大杜鹃和同属广布种马缨杜鹃的重测序数据分析显示, 朱红大杜鹃显著积累了更多的纯合有害突变, 这可能与朱红大杜鹃近交严重有关, 因为结果显示朱红大杜鹃的ROH程度显著高于马缨杜鹃。他们进一步注释了朱红大杜鹃中的有害突变, 检测到了几个与热应激相关的突变, 包括i热休克蛋白、ii转录因子(热应激转录因子、WRKY转录因子)和iii分子伴侣。朱红大杜鹃遗传多样性低, 遗传负荷重预示着该物种有很高的灭绝风险, 作者提出了一些相应的保护措施, 比如应当原地保护整个猴桥(HQ)种群, 禁止在该保护区进行任何活动以减缓该物种栖息地和野生种群的减少和退化; 设立永久的科学信息面板以提高公众对朱红大杜鹃的认识和保护意识。此外, 考虑到JT群体中有价值的基因型个体的灭绝, 他们提出应优先在当地附近的村庄和苗圃园进行全面调查找到某些具有解头(JT)群体遗传背景的个体。在这之后, 开展异地保护和人工补充授粉。Ma等(2022)基于重测序数据对漾濞槭不同种群的近交以及有害突变模式进行了分析, 制定出了个性化的遗传拯救模式。比如, 针对FROH高和纯合有害变异多的罗斯白地(LSBD)种群的遗传拯救, 他们建议利用纯合有害突变数目最低的打鹰山(DYS)种群个体的花粉来对LSBD种群的雌花进行授粉, 这样不但不会引入更多有害突变, 还可以使纯合的有害突变杂合化。另外, 崇仁(CR)种群由于遗传背景纯, FROH和有害突变较低也可以作为杂交的候选种群资源。
从以上例子可以看出, 全基因组重测序方法可以帮助鉴定近交衰退的遗传基础, 这些遗传信息可以用于自交衰退的早期诊断, 在设计育种计划时以帮助避免使用带有有害突变的个体, 因为一旦引入这样的个体将会影响野生种群的恢复。近交和有害突变的检测可以帮助制定出个性化的遗传拯救策略, 对于濒危植物的保护具有重要意义。
3 问题和展望
3.1 濒危植物保护存在的问题
伴随着基因组测序技术发展起来的保护基因组学为深入揭示保护生物学的问题带来了一些新的思路和解决办法, 逐渐成为保护生物学研究的重要手段。其中, 全基因组重测序方法是目前保护基因组学采用的组学方法中具有最高分辨力的一种方法, 其应用前景广阔。然而, 其广泛应用还存在以下问题: (1)大部分濒危物种高质量参考基因组缺乏, 测序成本较高, 和计算资源不足, 限制了全基因组重测序方法在保护生物学上的应用。(2)保护研究人员还需要相当熟练的生物信息学知识和技能。(3)保护基因组学研究和保护实践的应用之间仍然存在差距。
3.2 建议和未来发展趋势
保护基因组学不仅可以为传统保护遗传学关注的问题如遗传多样性、种群遗传结构等提供更强的统计支持, 而且还可以揭示种群大小随时间变化的动态历史, 更可以深入探讨物种或种群地方适应分子机制和近交衰退的遗传基础。而随着测序技术的进一步发展和测序成本的进一步下降, 可以预测未来会有越来越多濒危植物的基因组被解析出来, 且随着更多对用户友好的生物信息学工具和种群基因组学分析软件的开发, 基于全基因组重测序的保护基因组学研究面临的一些问题如缺少参考基因组的问题也必将迎刃而解, 重测序必将成为保护基因组学研究的主要技术手段。
在利用重测序方法研究保护生物学问题时, 我们建议: (1)研究者应与保护管理工作者建立专业良好的沟通体系, 确定要解决的问题, 并评估所要解决的保护生物学问题是否需要利用重测序方法才能解决, 如果利用传统的保护遗传学技术手段就可以解决的问题, 推荐优先利用传统的保护遗传学技术手段来解决; 但是当需要检测选择信号和确定表型性状或者适合度降低的遗传基础时, 全基因组重测序方法则是更好的选择。(2)管理建议应建立在取样充足和分析合理的基础上, 在确定重测序策略后, 进行群体取样时, 应尽可能将物种分布范围内的代表性群体全部取样, 在群体内部取样时, 个体之间的取样距离要足够; (3)获取重测序数据后需要利用多种软件对数据进行全面深入的分析, 比如目前就有多种软件可以进行种群动态的历史分析(如PSMC、Stairway plot2、SMC++和fastsimcoal2等软件), 对种群的适应性进化和有害突变研究也有多种不同的分析软件(前者有Fdist2和sweeperfinder等分析软件, 后者有SIFT和PolyPhen-2预测软件), 每种软件基于相似的或不同的原理, 可以帮助揭示物种进化的不同侧面, 通过比较多种分析结果有利于我们推断种群更加真实的演化历史和演化机制; (4)目前绝大多数基于重测序的保护基因组学研究都是基于核基因组的单核苷酸多态性SNP标记的分析, 对细胞器基因组标记的利用不足; 对于结构变异(structural variations, SV)如倒位(inversion)、异位(translocation)等在濒危物种演化过程中发挥的作用也知之甚少。随着长读长测序技术的进一步成熟, 研究人员可以更多地关注基因组中大的结构变异。(5)由于种群动态历史和遗传漂变形成的遗传模式也可能与地方性适应形成的遗传模式相似, 因此基因组扫描检测出来的位点需要进一步的功能验证。对表型性状和有适应效应的突变也需要设计实验进行证实以揭示其适应本质。建议一方面用模式物种来进行实验验证, 因为很多的生物途径在不同物种之间是保守的, 另一方面, 我们可以利用基因组编辑工具如CRISPR/Cas9在濒危植物中进行功能验证。
总之, 由于重测序方法可以帮助更加深入地窥探保护生物学的诸多问题, 它必将成为保护生物学研究新的常用的基本技术手段。
附录 Supplementary Material
附录1 非濒危植物种群核酸多样性的比较
Appendix 1 Comparison of nucleic acid diversity of non-threatened species groups
参考文献
A method and server for predicting damaging missense mutations
DOI:10.1038/nmeth0410-248 PMID:20354512 [本文引用: 1]
Genetics and the conservation of natural populations: Allozymes to genomes
DOI:10.1111/mec.13948
PMID:27933683
[本文引用: 1]

I consider how the study of genetic variation has influenced efforts to conserve natural populations over the last 50 years. Studies with allozymes in the 1970s provided the first estimates of the amount of genetic variation within and between natural populations at multiple loci. These early studies played an important role in developing plans to conserve species. The description of genetic variation in mitochondrial DNA in the early 1980s laid the foundation for the field of phylogeography, which provided a deeper look in time of the relationships and connectivity among populations. The development of microsatellites in the 1990s provided much more powerful means to describe genetic variation at nuclear loci, including the ability to detect past bottlenecks and estimate current effective population size with a single temporal sample. In the 2000s, single nucleotide polymorphisms presented a cornucopia of loci that has greatly improved power to estimate genetic and population demographic parameters important for conservation. Today, population genomics presents the ability to detect regions of the genome that are affected by natural selection (e.g. local adaptation or inbreeding depression). In addition, the ability to genotype historical samples has provided power to understand how climate change and other anthropogenic phenomena have affected populations. Modern molecular techniques provide unprecedented power to understand genetic variation in natural populations. Nevertheless, application of this information requires sound understanding of population genetics theory. I believe that current training in conservation genetics focuses too much on the latest techniques and too little on understanding the conceptual basis which is needed to interpret these data and ask good questions.© 2016 John Wiley & Sons Ltd.
Genomics and the future of conservation genetics
DOI:10.1038/nrg2844
PMID:20847747
[本文引用: 2]
We will soon have complete genome sequences from thousands of species, as well as from many individuals within species. This coming explosion of information will transform our understanding of the amount, distribution and functional significance of genetic variation in natural populations. Now is a crucial time to explore the potential implications of this information revolution for conservation genetics and to recognize limitations in applying genomic tools to conservation issues. We identify and discuss those problems for which genomics will be most valuable for curbing the accelerating worldwide loss of biodiversity. We also provide guidance on which genomics tools and approaches will be most appropriate to use for different aspects of conservation.
Harnessing the power of RADseq for ecological and evolutionary genomics
DOI:10.1038/nrg.2015.28
PMID:26729255
[本文引用: 3]
High-throughput techniques based on restriction site-associated DNA sequencing (RADseq) are enabling the low-cost discovery and genotyping of thousands of genetic markers for any species, including non-model organisms, which is revolutionizing ecological, evolutionary and conservation genetics. Technical differences among these methods lead to important considerations for all steps of genomics studies, from the specific scientific questions that can be addressed, and the costs of library preparation and sequencing, to the types of bias and error inherent in the resulting data. In this Review, we provide a comprehensive discussion of RADseq methods to aid researchers in choosing among the many different approaches and avoiding erroneous scientific conclusions from RADseq data, a problem that has plagued other genetic marker types in the past.
Perspective: Conservation genetics enters the genomics era
DOI:10.1007/s10592-009-0006-y URL [本文引用: 1]
Development of 85 SNP markers for the endangered plant species Prunus mira (Rosaceae) based on restriction site-associated DNA sequencing (RAD-seq)
DOI:10.1007/s12686-020-01140-0 [本文引用: 1]
Genome-wide association study reveals candidate genes involved in fruit trait variation in Persian walnut (Juglans regia L.)
DOI:10.3389/fpls.2020.607213
URL
[本文引用: 1]
Elucidating the genetic determinants of fruit quality traits in walnut is essential to breed new cultivars meeting the producers and consumers’ needs. We conducted a genome-wide association study (GWAS) using multi-locus models in a panel of 170 accessions of Juglans regia from the INRAE walnut germplasm collection, previously genotyped using the AxiomTMJ. regia 700K SNP array. We phenotyped the panel for 25 fruit traits related to morphometrics, shape, volume, weight, ease of cracking, and nutritional composition. We found more than 60 marker-trait associations (MTAs), including a highly significant SNP associated with nut face diameter, nut volume and kernel volume on chromosome 14, and 5 additional associations were detected for walnut weight. We proposed several candidate genes involved in nut characteristics, such as a gene coding for a beta-galactosidase linked to several size-related traits and known to be involved in fruit development in other species. We also confirmed associations on chromosomes 5 and 11 with nut suture strength, recently reported by the University of California, Davis. Our results enhance knowledge of the genetic control of important agronomic traits related to fruit quality in walnut, and pave the way for the development of molecular markers for future assisted selection.
Summary for policymakers of the global assessment report on biodiversity and ecosystem services of the Intergovernmental Science-Policy Platform on Biodiversity and Ecosystem Services
Genetic diversity of Horsfieldia tetratepala (Myristicaceae), an endangered Plant Species with Extremely Small Populations to China: Implications for its conservation
Vertebrates on the brink as indicators of biological annihilation and the sixth mass extinction
Liriodendron genome sheds light on angiosperm phylogeny and species-pair differentiation
DOI:10.1038/s41477-018-0323-6
[本文引用: 1]
The genus Liriodendron belongs to the family Magnoliaceae, which resides within the magnoliids, an early diverging lineage of the Mesangiospermae. However, the phylogenetic relationship of magnoliids with eudicots and monocots has not been conclusively resolved and thus remains to be determined1–6. Liriodendron is a relict lineage from the Tertiary with two distinct species—one East Asian (L. chinense (Hemsley) Sargent) and one eastern North American (L. tulipifera Linn)—identified as a vicariad species pair. However, the genetic divergence and evolutionary trajectories of these species remain to be elucidated at the whole-genome level7. Here, we report the first de novo genome assembly of a plant in the Magnoliaceae, L. chinense. Phylogenetic analyses suggest that magnoliids are sister to the clade consisting of eudicots and monocots, with rapid diversification occurring in the common ancestor of these three lineages. Analyses of population genetic structure indicate that L. chinense has diverged into two lineages—the eastern and western groups—in China. While L. tulipifera in North America is genetically positioned between the two L. chinense groups, it is closer to the eastern group. This result is consistent with phenotypic observations that suggest that the eastern and western groups of China may have diverged long ago, possibly before the intercontinental differentiation between L. chinense and L. tulipifera. Genetic diversity analyses show that L. chinense has tenfold higher genetic diversity than L. tulipifera, suggesting that the complicated regions comprising east–west-orientated mountains and the Yangtze river basin (especially near 30° N latitude) in East Asia offered more successful refugia than the south–north-orientated mountain valleys in eastern North America during the Quaternary glacial period.
Genomic analyses of a “living fossil”: The endangered dove-tree
DOI:10.1111/men.v20.3 URL [本文引用: 2]
Survival in the tropics despite isolation, inbreeding and asexual reproduction: Insights from the genome of the world’s southernmost poplar (Populus ilicifolia)
DOI:10.1111/tpj.v103.1 URL [本文引用: 1]
Rainbow: An integrated tool for efficient clustering and assembling RAD-seq reads
DOI:10.1093/bioinformatics/bts482
PMID:22942077
[本文引用: 1]
The innovation of restriction-site associated DNA sequencing (RAD-seq) method takes full advantage of next-generation sequencing technology. By clustering paired-end short reads into groups with their own unique tags, RAD-seq assembly problem is divided into subproblems. Fast and accurately clustering and assembling millions of RAD-seq reads with sequencing errors, different levels of heterozygosity and repetitive sequences is a challenging question.Rainbow is developed to provide an ultra-fast and memory-efficient solution to clustering and assembling short reads produced by RAD-seq. First, Rainbow clusters reads using a spaced seed method. Then, Rainbow implements a heterozygote calling like strategy to divide potential groups into haplotypes in a top-down manner. And along a guided tree, it iteratively merges sibling leaves in a bottom-up manner if they are similar enough. Here, the similarity is defined by comparing the 2nd reads of a RAD segment. This approach tries to collapse heterozygote while discriminate repetitive sequences. At last, Rainbow uses a greedy algorithm to locally assemble merged reads into contigs. Rainbow not only outputs the optimal but also suboptimal assembly results. Based on simulation and a real guppy RAD-seq data, we show that Rainbow is more competent than the other tools in dealing with RAD-seq data.Source code in C, Rainbow is freely available at http://sourceforge.net/projects/bio-rainbow/files/
Continuous base identification for single-molecule nanopore DNA sequencing
DOI:10.1038/nnano.2009.12
PMID:19350039
[本文引用: 1]
A single-molecule method for sequencing DNA that does not require fluorescent labelling could reduce costs and increase sequencing speeds. An exonuclease enzyme might be used to cleave individual nucleotide molecules from the DNA, and when coupled to an appropriate detection system, these nucleotides could be identified in the correct order. Here, we show that a protein nanopore with a covalently attached adapter molecule can continuously identify unlabelled nucleoside 5'-monophosphate molecules with accuracies averaging 99.8%. Methylated cytosine can also be distinguished from the four standard DNA bases: guanine, adenine, thymine and cytosine. The operating conditions are compatible with the exonuclease, and the kinetic data show that the nucleotides have a high probability of translocation through the nanopore and, therefore, of not being registered twice. This highly accurate tool is suitable for integration into a system for sequencing nucleic acids and for analysing epigenetic modifications.
Genomic signatures of evolution in Nautilus—An endangered living fossil
DOI:10.1111/mec.14344
PMID:28872211
[本文引用: 1]
Living fossils are survivors of previously more diverse lineages that originated millions of years ago and persisted with little morphological change. Therefore, living fossils are model organisms to study both long-term and ongoing adaptation and speciation processes. However, many aspects of living fossil evolution and their persistence in the modern world remain unclear. Here, we investigate three major aspects of the evolutionary history of living fossils: cryptic speciation, population genetics and effective population sizes, using members of the genera Nautilus and Allonautilus as classic examples of true living fossils. For this, we analysed genomewide ddRAD-Seq data for all six currently recognized nautiloid species throughout their distribution range. Our analyses identified three major allopatric Nautilus clades: a South Pacific clade, subdivided into three subclades with no signs of admixture between them; a Coral Sea clade, consisting of two genetically distinct populations with significant admixture; and a widespread Indo-Pacific clade, devoid of significant genetic substructure. Within these major clades, we detected five Nautilus groups, which likely correspond to five distinct species. With the exception of Nautilus macromphalus, all previously described species are at odds with genomewide data, testifying to the prevalence of cryptic species among living fossils. Detailed F analyses further revealed significant genome-wide and locus-specific signatures of selection between species and differentiated populations, which is demonstrated here for the first time in a living fossil. Finally, approximate Bayesian computation (ABC) simulations suggest large effective population sizes, which may explain the low levels of population differentiation commonly observed in living fossils.© 2017 John Wiley & Sons Ltd.
PyRAD: Assembly of de novo RADseq loci for phylogenetic analyses
DOI:10.1093/bioinformatics/btu121
PMID:24603985
[本文引用: 1]
Restriction-site-associated genomic markers are a powerful tool for investigating evolutionary questions at the population level, but are limited in their utility at deeper phylogenetic scales where fewer orthologous loci are typically recovered across disparate taxa. While this limitation stems in part from mutations to restriction recognition sites that disrupt data generation, an additional source of data loss comes from the failure to identify homology during bioinformatic analyses. Clustering methods that allow for lower similarity thresholds and the inclusion of indel variation will perform better at assembling RADseq loci at the phylogenetic scale.PyRAD is a pipeline to assemble de novo RADseq loci with the aim of optimizing coverage across phylogenetic datasets. It uses a wrapper around an alignment-clustering algorithm, which allows for indel variation within and between samples, as well as for incomplete overlap among reads (e.g. paired-end). Here I compare PyRAD with the program Stacks in their performance analyzing a simulated RADseq dataset that includes indel variation. Indels disrupt clustering of homologous loci in Stacks but not in PyRAD, such that the latter recovers more shared loci across disparate taxa. I show through reanalysis of an empirical RADseq dataset that indels are a common feature of such data, even at shallow phylogenetic scales. PyRAD uses parallel processing as well as an optional hierarchical clustering method, which allows it to rapidly assemble phylogenetic datasets with hundreds of sampled individuals.Software is written in Python and freely available at http://www.dereneaton.com/software/.© The Author 2014. Published by Oxford University Press. All rights reserved. For Permissions, please e-mail: journals.permissions@oup.com.
Genomics- informed delineation of conservation units in a desert amphibian
DOI:10.1111/mec.16660
PMID:35976166
[本文引用: 1]
Delineating conservation units (CUs, e.g., evolutionarily significant units, ESUs, and management units, MUs) is critical to the recovery of declining species because CUs inform both listing status and management actions. Genomic data have strengths and limitations in informing CU delineation and related management questions in natural systems. We illustrate the value of using genomic data in combination with landscape, dispersal and occupancy data to inform CU delineation in Nevada populations of the Great Basin Distinct Population Segment of the Columbia spotted frog (Rana luteiventris). R. luteiventris occupies naturally fragmented aquatic habitats in this xeric region, but beaver removal, climate change and other factors have put many of these populations at high risk of extirpation without management intervention. We addressed three objectives: (i) assessing support for ESUs within Nevada; (ii) evaluating and revising, if warranted, the current delineation of MUs; and (iii) evaluating genetic diversity, effective population size, adaptive differentiation and functional connectivity to inform ongoing management actions. We found little support for ESUs within Nevada but did identify potential revisions to MUs based on unique landscape drivers of connectivity that distinguish these desert populations from those in the northern portion of the species range. Effective sizes were uniformly small, with low genetic diversity and weak signatures of adaptive differentiation. Our findings suggest that management actions, including translocations and genetic rescue, might be warranted. Our study illustrates how a carefully planned genetic study, designed to address priority management goals that include CU delineation, can provide multiple insights to inform conservation action.© 2022 The Authors. Molecular Ecology published by John Wiley & Sons Ltd.
Testing for associations between loci and environmental gradients using latent factor mixed models
DOI:10.1093/molbev/mst063
PMID:23543094
[本文引用: 1]
Adaptation to local environments often occurs through natural selection acting on a large number of loci, each having a weak phenotypic effect. One way to detect these loci is to identify genetic polymorphisms that exhibit high correlation with environmental variables used as proxies for ecological pressures. Here, we propose new algorithms based on population genetics, ecological modeling, and statistical learning techniques to screen genomes for signatures of local adaptation. Implemented in the computer program "latent factor mixed model" (LFMM), these algorithms employ an approach in which population structure is introduced using unobserved variables. These fast and computationally efficient algorithms detect correlations between environmental and genetic variation while simultaneously inferring background levels of population structure. Comparing these new algorithms with related methods provides evidence that LFMM can efficiently estimate random effects due to population history and isolation-by-distance patterns when computing gene-environment correlations, and decrease the number of false-positive associations in genome scans. We then apply these models to plant and human genetic data, identifying several genes with functions related to development that exhibit strong correlations with climatic gradients.
Whole-genome sequencing approaches for conservation biology: Advantages, limitations and practical recommendations
DOI:10.1111/mec.14264
PMID:28746784
[本文引用: 9]
Whole-genome resequencing (WGR) is a powerful method for addressing fundamental evolutionary biology questions that have not been fully resolved using traditional methods. WGR includes four approaches: the sequencing of individuals to a high depth of coverage with either unresolved or resolved haplotypes, the sequencing of population genomes to a high depth by mixing equimolar amounts of unlabelled-individual DNA (Pool-seq) and the sequencing of multiple individuals from a population to a low depth (lcWGR). These techniques require the availability of a reference genome. This, along with the still high cost of shotgun sequencing and the large demand for computing resources and storage, has limited their implementation in nonmodel species with scarce genomic resources and in fields such as conservation biology. Our goal here is to describe the various WGR methods, their pros and cons and potential applications in conservation biology. WGR offers an unprecedented marker density and surveys a wide diversity of genetic variations not limited to single nucleotide polymorphisms (e.g., structural variants and mutations in regulatory elements), increasing their power for the detection of signatures of selection and local adaptation as well as for the identification of the genetic basis of phenotypic traits and diseases. Currently, though, no single WGR approach fulfils all requirements of conservation genetics, and each method has its own limitations and sources of potential bias. We discuss proposed ways to minimize such biases. We envision a not distant future where the analysis of whole genomes becomes a routine task in many nonmodel species and fields including conservation biology.© 2017 John Wiley & Sons Ltd.
Harnessing genomics for delineating conservation units
DOI:10.1016/j.tree.2012.05.012 URL [本文引用: 2]
Coming of age: Ten years of next-generation sequencing technologies
DOI:10.1038/nrg.2016.49
PMID:27184599
[本文引用: 1]
Since the completion of the human genome project in 2003, extraordinary progress has been made in genome sequencing technologies, which has led to a decreased cost per megabase and an increase in the number and diversity of sequenced genomes. An astonishing complexity of genome architecture has been revealed, bringing these sequencing technologies to even greater advancements. Some approaches maximize the number of bases sequenced in the least amount of time, generating a wealth of data that can be used to understand increasingly complex phenotypes. Alternatively, other approaches now aim to sequence longer contiguous pieces of DNA, which are essential for resolving structurally complex regions. These and other strategies are providing researchers and clinicians a variety of tools to probe genomes in greater depth, leading to an enhanced understanding of how genome sequence variants underlie phenotype and disease.
Research advances in conservation genetics and genomics of snow leopard (Panthera uncia)
雪豹保护遗传学和基因组学研究进展
DOI:10.16829/j.slxb.150687
[本文引用: 1]
雪豹 (Panthera uncia) 隶属于食肉目猫科豹属,是生活在青藏高原及其周边地区的旗舰物种。随着分子生物学和高通量测序技术的发展,雪豹保护遗传学和保护基因组学研究得到快速的发展,其中非损伤性遗传取样法显著推动了雪豹保护遗传学研究。本文综述了非损伤性遗传取样法在雪豹物种鉴定、个体识别和性别鉴定等研究中的应用,雪豹的系统发生地位、系统地理格局和种群遗传结构及其亚种争议、演化历史、适应性演化和基因组特征等保护遗传学和基因组学方面的研究现状和进展,并对雪豹保护遗传学和基因组学未来发展趋势进行了展望,以期促进雪豹保护生物学研究和保护对策的科学制定。
Population genomics for wildlife conservation and management
DOI:10.1111/mec.15720
PMID:33145846
[本文引用: 4]
Biodiversity is under threat worldwide. Over the past decade, the field of population genomics has developed across nonmodel organisms, and the results of this research have begun to be applied in conservation and management of wildlife species. Genomics tools can provide precise estimates of basic features of wildlife populations, such as effective population size, inbreeding, demographic history and population structure, that are critical for conservation efforts. Moreover, population genomics studies can identify particular genetic loci and variants responsible for inbreeding depression or adaptation to changing environments, allowing for conservation efforts to estimate the capacity of populations to evolve and adapt in response to environmental change and to manage for adaptive variation. While connections from basic research to applied wildlife conservation have been slow to develop, these connections are increasingly strengthening. Here we review the primary areas in which population genomics approaches can be applied to wildlife conservation and management, highlight examples of how they have been used, and provide recommendations for building on the progress that has been made in this field.© 2020 The Authors. Molecular Ecology published by 2020 John Wiley & Sons Ltd.
Two divergent haplotypes from a highly heterozygous lychee genome suggest independent domestication events for early and late-maturing cultivars
Genome evolution of the psammophyte Pugionium for desert adaptation and further speciation
Resequencing 93 accessions of coffee unveils independent and parallel selection during Coffea species divergence
DOI:10.1007/s11103-020-00974-4 [本文引用: 1]
Whole genome re-sequencing reveals evolutionary patterns of sacred lotus (Nelumbo nucifera)
DOI:10.1111/jipb.v60.1 URL [本文引用: 1]
A map of rice genome variation reveals the origin of cultivated rice
DOI:10.1038/nature11532 [本文引用: 1]
Targeted capture in evolutionary and ecological genomics
DOI:10.1111/mec.13304
PMID:26137993
[本文引用: 1]
The rapid expansion of next-generation sequencing has yielded a powerful array of tools to address fundamental biological questions at a scale that was inconceivable just a few years ago. Various genome-partitioning strategies to sequence select subsets of the genome have emerged as powerful alternatives to whole-genome sequencing in ecological and evolutionary genomic studies. High-throughput targeted capture is one such strategy that involves the parallel enrichment of preselected genomic regions of interest. The growing use of targeted capture demonstrates its potential power to address a range of research questions, yet these approaches have yet to expand broadly across laboratories focused on evolutionary and ecological genomics. In part, the use of targeted capture has been hindered by the logistics of capture design and implementation in species without established reference genomes. Here we aim to (i) increase the accessibility of targeted capture to researchers working in nonmodel taxa by discussing capture methods that circumvent the need of a reference genome, (ii) highlight the evolutionary and ecological applications where this approach is emerging as a powerful sequencing strategy and (iii) discuss the future of targeted capture and other genome-partitioning approaches in the light of the increasing accessibility of whole-genome sequencing. Given the practical advantages and increasing feasibility of high-throughput targeted capture, we anticipate an ongoing expansion of capture-based approaches in evolutionary and ecological research, synergistic with an expansion of whole-genome sequencing.© 2015 John Wiley & Sons Ltd.
A spatial analysis method (SAM) to detect candidate loci for selection: Towards a landscape genomics approach to adaptation
The detection of adaptive loci in the genome is essential as it gives the possibility of understanding what proportion of a genome or which genes are being shaped by natural selection. Several statistical methods have been developed which make use of molecular data to reveal genomic regions under selection. In this paper, we propose an approach to address this issue from the environmental angle, in order to complement results obtained by population genetics. We introduce a new method to detect signatures of natural selection based on the application of spatial analysis, with the contribution of geographical information systems (GIS), environmental variables and molecular data. Multiple univariate logistic regressions were carried out to test for association between allelic frequencies at marker loci and environmental variables. This spatial analysis method (SAM) is similar to current population genomics approaches since it is designed to scan hundreds of markers to assess a putative association with hundreds of environmental variables. Here, by application to studies of pine weevils and breeds of sheep we demonstrate a strong correspondence between SAM results and those obtained using population genetics approaches. Statistical signals were found that associate loci with environmental parameters, and these loci behave atypically in comparison with the theoretical distribution for neutral loci. The contribution of this new tool is not only to permit the identification of loci under selection but also to establish hypotheses about ecological factors that could exert the selection pressure responsible. In the future, such an approach may accelerate the process of hunting for functional genes at the population level.
Earth BioGenome Project: Sequencing life for the future of life
Advances in plant conservation genetics
DOI:10.17520/biods.2002009
[本文引用: 1]
Conservation genetics is a new field of research focusing on the studies and practices of biodiversity conservation based on the principles and techniques of population genetics. During the past decades, genetic studies have made increasingly great contributions to biodiversity conservation in theory and practice. In this paper, we briefly introduce the concept and history of conservation genetics, and highlight progress in plant conservation genetics. Four major aspects of conservation genetics in plants are addressed, including plant phylogenetic reconstruction and identification of conservation units, the relationship between genetic diversity and species fitness, population genetic structure and conservation strategies, as well as the identification and utilization of plant genetic resources. In addition, the great importance of genetic studies in plant conservation is discussed.
植物保护遗传学研究进展
DOI:10.17520/biods.2002009
[本文引用: 1]
保护遗传学是运用遗传学的原理和研究手段,以生物多样性尤其是遗传多样性的研究和保护为核心的一门新兴学科。近几十年来,遗传学研究在生物多样性保护的理论和实践中发挥着越来越重要的作用。本文简要回顾了保护遗传学的发展历史、研究方向和涉及的概念,着重介绍了植物保护遗传学研究所取得的一些进展,包括植物系统发育重建和保护单元的确定、遗传多样性与物种和群体适应性之间的关系、群体遗传结构与保护策略的制定以及植物遗传资源的鉴定和利用等方面的内容,并强调保护遗传学研究是未来生物多样性和保护生物学研究中一个亟待加强的研究领域。
The genome of the Paleogene relic tree Bretschneidera sinensis: Insights into trade-offs in gene family evolution, demographic history, and adaptive SNPs
DOI:10.1093/dnares/dsac003
URL
[本文引用: 4]
Among relic species, genomic information may provide the key to inferring their long-term survival. Therefore, in this study, we investigated the genome of the Paleogene relic tree species, Bretschneidera sinensis, which is a rare endemic species within southeastern Asia. Specifically, we assembled a high-quality genome for B. sinensis using PacBio high-fidelity and high-throughput chromosome conformation capture reads and annotated it with long and short RNA sequencing reads. Using the genome, we then detected a trade-off between active and passive disease defences among the gene families. Gene families involved in salicylic acid and MAPK signalling pathways expanded as active defence mechanisms against disease, but families involved in terpene synthase activity as passive defences contracted. When inferring the long evolutionary history of B. sinensis, we detected population declines corresponding to historical climate change around the Eocene–Oligocene transition and to climatic fluctuations in the Quaternary. Additionally, based on this genome, we identified 388 single nucleotide polymorphisms (SNPs) that were likely under selection, and showed diverse functions in growth and stress responses. Among them, we further found 41 climate-associated SNPs. The genome of B. sinensis and the SNP dataset will be important resources for understanding extinction/diversification processes using comparative genomics in different lineages.
The Tetracentron genome provides insight into the early evolution of eudicots and the formation of vessel elements
DOI:10.1186/s13059-020-02198-7
[本文引用: 4]
Tetracentron sinense is an endemic and endangered deciduous tree. It belongs to the Trochodendrales, one of four early diverging lineages of eudicots known for having vesselless secondary wood. Sequencing and resequencing of the T. sinense genome will help us understand eudicot evolution, the genetic basis of tracheary element development, and the genetic diversity of this relict species.
Application of genomics technology in biodiversity conservation research
DOI:10.17520/biods.2022441
[本文引用: 2]
Background: Research techniques in molecular biology, cell biology, microbiology and genetics have been accelerated by rapid development of modern genomic technologies. These advances have rapidly evolved the field of biodiversity research, once a branch of natural history focusing on morphology, into an integrated life science. Modern biodiversity studies can now investigate and link element of ecological systems, the species within them, and their genetic diversity. DNA related technologies, among other omics techniques, have continued to develop and launch new sequencing platforms, leading to a reduction of DNA sequencing costs that has already outstripped Moore’s Law, which also facilitates a series of breakthroughs in the research fields of biodiversity. Prospects: Here, we introduce emerging trends in DNA-based omics techniques applied in biodiversity research, including species-level genomics as well as genetic diversity and community-levels species diversity. The former includes genomes obtained based on single individuals and genetic diversity of focal populations in both spatial and temporal dimensions, while the latter includes molecular identification approaches, such as metabarcoding, eDNA, iDNA etc. These new methods can be applied in biodiversity estimation for various communities, as well as in monitoring and conservation of flagship species and interspecific interactions.
基因组学技术在生物多样性保护研究中的应用
DOI:10.17520/biods.2022441
[本文引用: 2]
在分子生物学、细胞生物学、微生物学、遗传学等学科的推动下, 生物多样性研究从仅关注宏观表型的博物学, 迅速演化为涵盖生态系统、物种和遗传多样性等多个维度的综合性生命科学。组学技术, 尤其是DNA测序技术的更新和发展, 使获取DNA序列所需的成本大幅下降, 促进了近年来其在生物多样性研究中取得的一系列令人瞩目成就。本文将从物种水平的遗传多样性和群落水平的物种多样性两个层面总结和介绍与DNA相关的组学技术在生物多样性研究中的一些创新和应用。其中, 物种水平主要是总结单一个体的基因组和单物种多个体在时空多个维度上的群体遗传研究; 而群落水平的物种多样性层面主要总结现有的分子鉴定技术(metabarcoding, eDNA, iDNA等), 以及上述新技术在群落多样性评估、旗舰保护物种监测以及物种间相互作用关系等研究中的应用。
Genome-level diversification of eight ancient tea populations in the Guizhou and Yunnan regions identifies candidate genes for core agronomic traits
DOI:10.1038/s41438-021-00617-9
PMID:34376642
[本文引用: 2]
The ancient tea plant, as a precious natural resource and source of tea plant genetic diversity, is of great value for studying the evolutionary mechanism, diversification, and domestication of plants. The overall genetic diversity among ancient tea plants and the genetic changes that occurred during natural selection remain poorly understood. Here, we report the genome resequencing of eight different groups consisting of 120 ancient tea plants: six groups from Guizhou Province and two groups from Yunnan Province. Based on the 8,082,370 identified high-quality SNPs, we constructed phylogenetic relationships, assessed population structure, and performed genome-wide association studies (GWAS). Our phylogenetic analysis showed that the 120 ancient tea plants were mainly clustered into three groups and five single branches, which is consistent with the results of principal component analysis (PCA). Ancient tea plants were further divided into seven subpopulations based on genetic structure analysis. Moreover, it was found that the variation in ancient tea plants was not reduced by pressure from the external natural environment or artificial breeding (nonsynonymous/synonymous = 1.05). By integrating GWAS, selection signals, and gene function prediction, four candidate genes were significantly associated with three leaf traits, and two candidate genes were significantly associated with plant type. These candidate genes can be used for further functional characterization and genetic improvement of tea plants.© 2021. The Author(s).
Chromosome-level genome assembly and population genetic analysis of a critically endangered Rhododendron provide insights into its conservation
DOI:10.1111/tpj.v107.5 URL [本文引用: 4]
Demographic history and identification of threats revealed by population genomic analysis provide insights into conservation for an endangered maple
DOI:10.1111/mec.v31.3 URL [本文引用: 2]
Genome-wide analysis of butterfly bush (Buddleja alternifolia) in three uplands provides insights into biogeography, demography and speciation
DOI:10.1111/nph.v232.3 URL [本文引用: 2]
Assessing habitat suitability for the translocation of Ochrosia tahitensis (Apocynaceae), a critically endangered endemic plant from the island of Tahiti (South Pacific)
DOI:10.1016/j.jnc.2022.126198 URL [本文引用: 1]
SIFT: Predicting amino acid changes that affect protein function
DOI:10.1093/nar/gkg509
PMID:12824425
[本文引用: 1]
Single nucleotide polymorphism (SNP) studies and random mutagenesis projects identify amino acid substitutions in protein-coding regions. Each substitution has the potential to affect protein function. SIFT (Sorting Intolerant From Tolerant) is a program that predicts whether an amino acid substitution affects protein function so that users can prioritize substitutions for further study. We have shown that SIFT can distinguish between functionally neutral and deleterious amino acid changes in mutagenesis studies and on human polymorphisms. SIFT is available at http://blocks.fhcrc.org/sift/SIFT.html.
The chromosome-level reference genome assembly for Dendrobium officinale and its utility of functional genomics research and molecular breeding study
DOI:10.1016/j.apsb.2021.01.019 URL [本文引用: 1]
From conservation genetics to conservation genomics
RNA-Seq and its applications: A new technology for transcriptomics
DOI:10.3724/SP.J.1005.2011.01191 URL [本文引用: 1]
转录组研究新技术: RNA-Seq及其应用
Next-generation sequencing transforms today’s biology
DOI:10.1038/nmeth1156
PMID:18165802
[本文引用: 1]
A new generation of non-Sanger-based sequencing technologies has delivered on its promise of sequencing DNA at unprecedented speed, thereby enabling impressive scientific achievements and novel biological applications. However, before stepping into the limelight, next-generation sequencing had to overcome the inertia of a field that relied on Sanger-sequencing for 30 years.
The evolutionary truth about living fossils
DOI:10.1511/2014.111.434 URL [本文引用: 1]
From conservation genetics to conservation genomics
从保护遗传学到保护基因组学
Interpretation of variation across marker loci as evidence of selection
DOI:10.1093/genetics/158.4.1811
PMID:11514464
[本文引用: 1]
Population structure and history have similar effects on the genetic diversity at all neutral loci. However, some marker loci may also have been strongly influenced by natural selection. Selection shapes genetic diversity in a locus-specific manner. If we could identify those loci that have responded to selection during the divergence of populations, then we may obtain better estimates of the parameters of population history by excluding these loci. Previous attempts were made to identify outlier loci from the distribution of sample statistics under neutral models of population structure and history. Unfortunately these methods depend on assumptions about population structure and history that usually cannot be verified. In this article, we define new population-specific parameters of population divergence and construct sample statistics that are estimators of these parameters. We then use the joint distribution of these estimators to identify outlier loci that may be subject to selection. We found that outlier loci are easier to recognize when this joint distribution is conditioned on the total number of allelic states represented in the pooled sample at each locus. This is so because the conditional distribution is less sensitive to the values of nuisance parameters.
Development of microsatellite markers for a monotypic and globally endangered species, Glyptostrobus pensilis (Cupressaceae)
Whole-genome resequencing reveals signature of local adaptation and divergence in wild soybean
DOI:10.1111/eva.13480
PMID:36426120
[本文引用: 4]
Global climate change has threatened world crop production and food security. Decoding the adaptive genetic basis of wild relatives provides an invaluable genomic resource for climate-smart crop breedinG. Here, we performed whole-genome sequencing of 185 diverse wild soybean () accessions collected from three major agro-ecological zones in China to parse the genomic basis of local adaptation in wild soybean. The population genomic diversity pattern exhibited clear agro-ecological zone-based population structure, and multiple environmental factors were observed to contribute to the genetic divergence. Demographic analysis shows that wild soybeans from the three ecological zones diverged about 1 × 10 years ago, and then the effective population sizes have undergone different degrees of expansions. Genome-environment association identified multiple genes involved in the local adaptation, such as flowering time and temperature-related genes. A locus containing two adjacent MADS-box transcription factors on chromosome 19 was identified for multiple environmental factors, and it experienced positive selection that enables the adaptation to high-latitude environment. This study provides insights into the genetic mechanism of ecological adaptation in wild soybean that may facilitate climate-resilient soybean breeding.© 2022 The Authors. Evolutionary Applications published by John Wiley & Sons Ltd.
Exome sequencing: Current and future perspectives
Research advances and perspectives of conservation genomics and meta-genomics of threatened mammals in China
DOI:10.16829/j.slxb.150518
[本文引用: 3]
Understanding the evolutionary processes, endangered mechanisms and adaptive evolution are key scientific issues in conservation biology. During the past decades, advances in high-throughput sequencing and multi-disciplinary crossover provide deep insights into the evolutionary history, genetic structure, adaptive evolution, and host-microbiota coevolution of endangered species. The emergence of two new branches of conservation biology, Conservation Genomics and Conservation Metagenomics, provides novel insights into wildlife conservation. In this review, we summarize the important advances in the two fields and discuss the future research directions, aiming to promote the conservation biology of threa-tened animals in China.
中国濒危兽类保护基因组学和宏基因组学研究进展与展望
DOI:10.16829/j.slxb.150518
[本文引用: 3]
揭示濒危兽类的演化历史、濒危机制及适应性演化策略,是保护生物学关注的重大科学问题。近十几年来,高通量测序技术的不断发展以及多学科交叉融合,为揭示濒危物种的演化历史、遗传结构、适应性演化及其与肠道微生物的协同演化的分子机制提供了重要的技术支撑,由此产生了保护基因组学和保护宏基因组学两个分支学科,为野生动物尤其是濒危动物的保护生物学研究提供了新的方向与思路。本文综述了我国在保护基因组学和保护宏基因组学领域取得的重要进展,并展望未来的发展趋势,以期进一步推动我国濒危兽类保护生物学的发展。
Chromosome- level de novo genome assembly and whole-genome resequencing of the threatened species Acanthochlamys bracteata (Velloziaceae) provide insights into alpine plant divergence in a biodiversity hotspot
DOI:10.1111/men.v22.4 URL [本文引用: 1]
Genomic effects of population collapse in a critically endangered ironwood tree Ostrya rehderiana
DOI:10.1038/s41467-018-07913-4
[本文引用: 4]
Increased human activity and climate change are driving numerous tree species to endangered status, and in the worst cases extinction. Here we examine the genomic signatures of the critically endangered ironwood tree Ostrya rehderiana and its widespread congener O. chinensis. Both species have similar demographic histories prior to the Last Glacial Maximum (LGM); however, the effective population size of O. rehderiana continued to decrease through the last 10,000 years, whereas O. chinensis recovered to Pre-LGM numbers. O. rehderiana accumulated more deleterious mutations, but purged more severely deleterious recessive variations than in O. chinensis. This purging and the gradually reduced inbreeding depression together may have mitigated extinction and contributed to the possible future survival of the outcrossing O. rehderiana. Our findings provide critical insights into the evolutionary history of population collapse and the potential for future recovery of the endangered trees.
Resequencing 545 ginkgo genomes across the world reveals the evolutionary history of the living fossil
DOI:10.1038/s41467-019-12133-5
[本文引用: 5]
As Charles Darwin anticipated, living fossils provide excellent opportunities to study evolutionary questions related to extinction, competition, and adaptation. Ginkgo (Ginkgo biloba L.) is one of the oldest living plants and a fascinating example of how people have saved a species from extinction and assisted its resurgence. By resequencing 545 genomes of ginkgo trees sampled from 51 populations across the world, we identify three refugia in China and detect multiple cycles of population expansion and reduction along with glacial admixture between relict populations in the southwestern and southern refugia. We demonstrate multiple anthropogenic introductions of ginkgo from eastern China into different continents. Further analyses reveal bioclimatic variables that have affected the geographic distribution of ginkgo and the role of natural selection in ginkgo’s adaptation and resilience. These investigations provide insights into the evolutionary history of ginkgo trees and valuable genomic resources for further addressing various questions involving living fossil species.
Genomic insights on the contribution of balancing selection and local adaptation to the long-term survival of a widespread living fossil tree, Cercidiphyllum japonicum
DOI:10.1111/nph.v228.5 URL [本文引用: 2]

{kind=link}
{kind=link}
{kind=link}
{kind=link}