



福寿螺(Gastropoda: Ampullariidae: Pomacea)是原产于南美洲亚马孙流域的一类大型淡水螺(Cowie et al, 2006), 当前已扩散至亚洲、北美洲、欧洲、大洋洲的30余个国家和地区(Brito & Joshi, 2016)。在我国, 福寿螺最早以食用为目的引入到台湾养殖, 随后引入到广东省中山市, 并在20世纪80年代的养殖热中在全国迅速扩散(杨叶欣等, 2010; Yang et al, 2018)。由于福寿螺携带广州管圆线虫(Angiostrongylus cantonensis)等多种高风险人畜共患寄生虫, 导致其逐渐失去市场价值, 被大量弃养并逃逸到野外(Hayes et al, 2008)。迄今福寿螺已在我国南方17个省区市(海南、广东、广西、云南、贵州、福建、江西、四川、重庆、湖南、湖北、安徽、浙江、江苏、上海、香港、台湾)广泛扩散, 对入侵地的农业生产和人体健康造成严重威胁(Yang et al, 2018, 2022; 戢小梅等, 2020; 刘义满等, 2020)。此外, 福寿螺食性杂, 通过牧食淡水植物、捕食淡水无脊椎动物及底栖生物、竞争淡水水生资源等对水体食物链结构、生物多样性产生系列损害, 严重威胁水生生态安全。
尽管人们对于福寿螺的入侵危害已有较深刻的认识, 但在很长时间内对福寿螺的种类及分布并不清楚。福寿螺属的许多物种形态近似、可塑性高, 根据螺壳等形态学特征很难准确鉴别种类(Hayes et al, 2012)。早期国内外普遍认为入侵福寿螺仅包括小管福寿螺(Pomacea canaliculata) (Hayes et al, 2015), 近年来, 利用母系遗传的线粒体基因, 通过序列相似度比对、系统发育关系分析、序列特征分析等(刘青青和董志军, 2018), 可以实现福寿螺种类的有效鉴定(杨倩倩等, 2016), 从而为福寿螺的分类提供了有力支撑。例如, Yang等(2019)基于线粒体细胞色素c氧化酶亚基I (cytochrome c oxidase subunit I, COI)基因序列的DNA条形码分析从我国分布的福寿螺种群中鉴别出小管福寿螺、斑点福寿螺(Pomacea maculata), 并发现1个隐存种; 进一步结合螺壳、软体解剖特征、卵及幼螺形态等的系统研究, 将这一隐存种命名为隐秘福寿螺(Pomacea occulta) (Yang & Yu, 2019)。当前已明确小管福寿螺在我国分布范围最广, 在南方各省份均有分布; 隐秘福寿螺已扩散至13省份; 斑点福寿螺分布于四川盆地、浙江、江苏与香港(Yang et al, 2018, 2022; 钱子衿等, 2021)。
受气候变化、经济一体化等影响, 福寿螺在全球的扩散趋势日益加剧。Lei等(2017)根据物种分布模型(SDMs)分析, 发现限制小管福寿螺在全球扩散的最主要气候因子是最寒冷月份的最低气温, 推测在今后50年内小管福寿螺将在亚洲和北美洲向北、欧洲向东进一步扩散, 全球适生区将增加3.3%-10.3%。我国学者以多种气候因子为基础, 针对福寿螺的分布北界和扩散趋势开展了系列研究。Lv等(2011)首次以环境温度与小管福寿螺世代构建模型并结合GIS绘图, 预测2020年小管福寿螺在我国的最北分布范围可达31° N; 杨海芳等(2018)以气温和降水量等作为气候影响因子, 预测福寿螺在我国的适生区北界为35° N; Yin等(2022)研究发现, 最暖季度的降水量和最冷月份的最高气温对小管福寿螺在我国的分布起重要作用, 随着全球气候变暖, 小管福寿螺未来将会进一步向我国北方地区扩散。近年来, 福寿螺在江苏及上海地区大面积发生, 在农田等水生生态系统中为害严重, 引起广泛关注(Yang et al, 2019)。田间调研发现, 2019年福寿螺分布最北已达32.43° N的江苏省泰州市(戢小梅等, 2020)。
此外, 福寿螺种间渐渗杂交的发现成为其入侵机制研究的新热点。Matsukura等(2013)首次利用限制性内切酶ApaLI对核EF1α基因的酶切图谱多态性进行分析, 发现日本、韩国、菲律宾、越南的小管福寿螺和斑点福寿螺发生了种间杂交渐渗。Yang等(2020)构建了检测福寿螺杂交型的特异引物多重PCR方法, 检测了采集自中国12省份的福寿螺, 结果表明线粒体为小管福寿螺、核基因为小管福寿螺和斑点福寿螺杂合型的杂交种在我国分布最广泛, 并揭示了杂交种的卵形态, 如卵块大小和卵粒直径介于两纯种福寿螺之间。此外, 在原产地阿根廷、巴西、乌拉圭等也检测到小管福寿螺和斑点福寿螺的种间渐渗杂交(Yoshida et al, 2014; Glasheen et al, 2020)。通过实验室交配试验, 揭示了小管福寿螺比斑点福寿螺具有更高的耐低温能力, 而二者的杂交种耐低温水平介于两个纯种之间, 这与日本田间调研的结果一致, 即杂交种福寿螺在日本分布的纬度范围介于两纯种之间, 推测渐渗杂交有助于提升斑点福寿螺的耐寒性, 促进其向低温地区扩散(Matsukura et al, 2013, 2016)。
本研究从上海及江苏跨长江分布区广泛采集了福寿螺样品, 利用线粒体COI基因序列进行种类鉴定, 结合入侵地与原产地种群已发表的COI序列进行种群遗传结构和遗传多样性分析, 并通过核EF1α基因分型分析探明该地区福寿螺种群的种间渐渗杂交情况。
1 材料与方法
1.1 样品采集
2020年10月至2021年10月采集长江下游11个地理种群的福寿螺样本。其中, 长江下游以南地区有8个种群, 即上海市宝山区种群(SHBS)、太湖沿岸的苏州市吴中区(WZLZ、WZMJB)、昆山市周庄镇(KSZZ)和常熟市尚湖镇(CSSH) 4个种群, 以及临近长江南岸的常熟市古里镇(CSGL)和张家港市(ZJGFH、ZJGXSC)种群; 长江下游以北包括扬州市广陵区(JSYZ)、泰州市兴化市(JSTZ)和宿迁市泗洪县(JSSQ) 3个种群(表1, 图1)。其中, 宿迁市泗洪县(118.25° E, 33.47° N)为最北的采样点。上海种群及长江下游以北的种群主要采自河道, JSTZ除灌溉河道外还包括蟹田, JSSQ种群采自湖泊, ZJGFH种群采自沟渠, 其他种群采自稻田或水生蔬菜田。
表1 长江下游分布区福寿螺样品采集信息
Table 1
| 序号 Code | 地点 Locality | 编号 Code | 经度 Longitude (E) | 纬度 Latitude (N) | 生境 Habitat | 序列数量 No. of sequences |
|---|---|---|---|---|---|---|
| 1 | 上海市宝山区三星村 Sanxing Village, Baoshan District, Shanghai | SHBS | 121.36° | 31.31° | 河道 River | 20 |
| 2 | 苏州市吴中区甪直镇 Luzhi Town, Wuzhong District, Suzhou City | WZLZ | 120.48° | 31.14° | 水生蔬菜田 Aquatic vegetable field | 30 |
| 3 | 苏州市吴中区马家浜村 Majiabang Village, Wuzhong District, Suzhou City | WZMJB | 120.46° | 31.14° | 水生蔬菜田 Aquatic vegetable field | 30 |
| 4 | 昆山市周庄镇 Zhouzhuang Town, Kunshan City | KSZZ | 120.56° | 31.21° | 稻田 Paddy field | 30 |
| 5 | 常熟市尚湖镇 Shanghu Town, Changshu City | CSSH | 120.42° | 31.36° | 稻田 Paddy field | 30 |
| 6 | 常熟市古里镇 Guli Town, Changshu City | CSGL | 120.85° | 31.65° | 稻田 Paddy field | 15 |
| 7 | 张家港市凤凰镇鸷山 Fenghuang Town, Zhangjiagang City | ZJGFH | 120.62° | 31.76° | 沟渠 Ditch | 17 |
| 8 | 张家港市凤凰镇杏市村 Xingshi Village, Fenghuang Town, Zhangjiagang City | ZJGXSC | 120.68° | 31.78° | 稻田 Paddy field | 28 |
| 9 | 扬州市广陵区沙头镇 Shatou Town, Guangling District, Yangzhou City | JSYZ | 119.50° | 32.34° | 河道 River | 30 |
| 10 | 泰州市兴化市大邹镇顾马村 Guma Village, Dazou Town, Xinghua City, Taizhou City | JSTZ | 119.92° | 33.15° | 河道、蟹田 River, crab field | 30 |
| 11 | 宿迁市泗洪县 Sihong County, Suqian City | JSSQ | 118.25° | 33.47° | 湖泊 Lake | 10 |
图1
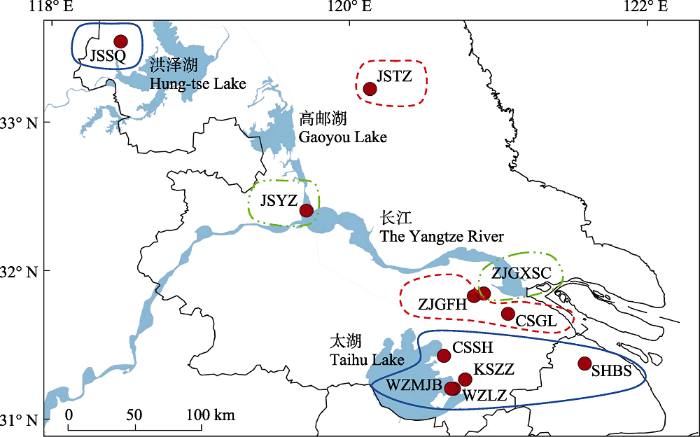
图1
本研究福寿螺采样点及基于AMOVA层次分析的分组。实心圆表示采样点, 同一线型框表示同一组群, 图中缩写含义见
Fig. 1
Sampling locations of the apple snails and grouping based on AMOVA hierarchical analysis. Solid circles represent sampling points, the frames in same linetype indicates a population group, the meaning of the abbreviation in the figure is shown in
所用样品中, 张家港市凤凰镇(ZJGFH)和宿迁市泗洪县(JSSQ)样点分别包含13个和10个卵块, 其余均为成螺。其中, 采集样本量多于30个的种群从中随机抽取30个, 样本不足30个的种群取全部, 共计270个样本用于分子实验。
1.2 DNA提取、PCR扩增及测序
取约10 mg成螺腹足或单粒卵提取基因组DNA, 步骤参考血液/细胞/组织基因组DNA提取试剂盒(TIANGEN, 上海)说明书。每个样品获得的DNA溶于100 µL ddH2O中, 使用微量紫外分光光度仪(NanoDrop, ND-2000)检测浓度及纯度后, 置于-20℃保存。
使用通用引物对LCO1490/HCO2198 (Folmer et al, 1994)扩增线粒体COI基因片段。PCR体系包括12.5 µL的TaKaRa Premix Taq, 9.5 µL的ddH2O, 上下游引物(10 µM)各1 µL以及1 µL的基因组DNA。PCR反应条件为: 94℃预变性3 min; 94℃变性30 s, 50℃退火30 s, 72℃延伸1 min, 共34个循环; 72℃延伸10 min, 4℃终止反应。使用1%的琼脂糖凝胶对PCR产物进行电泳检测, 将获得的阳性扩增产物交由上海生工生物工程股份有限公司纯化测序。使用Geneious v11.1.5 (Kearse et al, 2012)软件对原始序列进行人工校正并剪切两端引物后获得最终序列。
1.3 COI数据集
我们获取了文献中已发表的中国大陆和香港以及日本、巴西和阿根廷种群的福寿螺COI序列702条, 与本研究所测的270条序列形成包含972条序列的数据集。其中, 中国大陆种群包括319条小管福寿螺序列、30条斑点福寿螺序列(Yang et al, 2018), 以及68条隐秘福寿螺序列(Yang et al, 2019); 香港种群分别包括小管福寿螺、斑点福寿螺和隐秘福寿螺序列139条、10条和13条(Yang et al, 2022); 日本种群的19条小管福寿螺序列和29条斑点福寿螺序列(Matsukura et al, 2008); 阿根廷种群的32条小管福寿螺序列和13条斑点福寿螺序列(Rawlings et al, 2007; Hayes et al, 2008, 2009); 巴西种群的斑点福寿螺序列30条(Hayes et al, 2008; Glasheen et al, 2020) (附录2)。其中, 中国大陆、香港以及日本种群的序列为地理种群序列, 而阿根廷和巴西种群的序列主要是单倍型序列。
1.4 序列相似性与系统发育分析
以神秘福寿螺(Pomacea diffusa) (EF515067)作为外群, 基于相邻连接法(neighbor-joining, NJ)和贝叶斯法(Bayesian inference, BI)构建系统发育树。使用MEGA X软件(Kagawa & Takimoto, 2018)基于K2P模型构建NJ树, 置信值设置为1,000次; 使用jModeltest v2.1.10中的BIC计算出最佳核苷酸替代模型为HKY + G, 并使用MrBayes v3.2.5软件(Ronquist & Huelsenbeck, 2003)构建BI树, 同时运行4个马尔可夫链(MCMC), 共运算5,000,000代, 每1,000代储存一次树, 用burnin参数舍弃前25%后, 计算合一树。
1.5 种群遗传结构和种群遗传多样性分析
在鉴定福寿螺物种的基础上, 对COI数据集生成的小管福寿螺和斑点福寿螺单倍型进行种群遗传结构分析。使用TCS v1.21 (Clement et al, 2000)对数据集进行单倍型网络分析, 网络重建的统计简约连接界限(parsimony limit)设置为95%。
针对本研究所测定的长江下游江苏和上海种群、已发表的中国大陆种群及香港种群的小管福寿螺和斑点福寿螺进行种群遗传多样性比较分析。使用DnaSP v5.1 (Rozas et al, 2017)计算小管福寿螺和斑点福寿螺的单倍型多样性(haplotype diversity, Hd), 核苷酸多样性(nucleotide diversity, π)和平均核苷酸差异数(average number of nucleotide difference, k)等遗传多样性参数。对江苏和上海地区长江南、北小管福寿螺种群进行了遗传多样性比较分析, 以揭示该地区长江南北福寿螺种群遗传多样性的关系。
为了揭示江苏和上海地区长江南北福寿螺种群的关系, 使用SAMOVA v2.0软件(Excoffier et al, 1992)对11个地理种群的小管福寿螺进行AMOVA层次分析, 设置分组为2‒7组, 采用标准计算中的Tamura & Nei模型进行1,023个置换检验, 计算11个地理种群的分组以及各种群变异来源和变异程度。
1.6 杂交渐渗分析
使用Yang等(2020)发表的特异引物多重PCR方法, 利用引物组EF3Fc/EF3Rc/EF1Fm/EF3Rm进行核基因EF1α分型分析。使用3.5% (w/v)琼脂糖凝胶电泳检测3 μL的扩增产物。
根据线粒体COI基因(C、M型)和核基因EF1α (C、M、B型)共同判断福寿螺个体的杂交型。其中, 线粒体COI基因鉴定为小管福寿螺时, 核基因EF1α也鉴定为小管福寿螺的是CC型; 核基因EF1α鉴定为斑点福寿螺的是CM型; 核基因EF1α鉴定为小管福寿螺和斑点福寿螺杂合型的是CB型。同样地, 线粒体COI基因鉴定为斑点福寿螺时, 根据不同的核基因EF1α分型可分为MC型、MM型和MB型3种杂交型。
2 结果
2.1 福寿螺物种鉴定
本研究共测序获得长江下游江苏和上海种群福寿螺的270条COI序列(GenBank序列号MZ396656-MZ396815, ON054061-ON054170)。所构建的包含中国大陆和香港以及日本、巴西和阿根廷福寿螺种群的972条COI序列数据集, 经序列多重比对后剪切为579 bp的一致性序列, 并生成73个单倍型(Hap1-73, 附录3)。所测长江下游江苏和上海种群的福寿螺COI序列分布于单倍型Hap1-10。
依据序列相似性分析73个单倍型聚类为3组(图2)。其中, 第一组共包含30个单倍型, Hap1-9和小管福寿螺单倍型Hap11-31, 组内遗传距离为94.0%‒ 99.8%, 组间遗传距离为89.0%‒92.8%; 第二组包括38个单倍型, Hap10和斑点福寿螺单倍型Hap32-68, 组内遗传距离为94.8%‒99.8%, 组间遗传距离为89.0%‒94.3%; 第三组包含隐秘福寿螺的单倍型Hap69-73, 组内遗传距离为99.1%‒99.8%, 组间遗传距离为89.6%‒94.3%。
图2
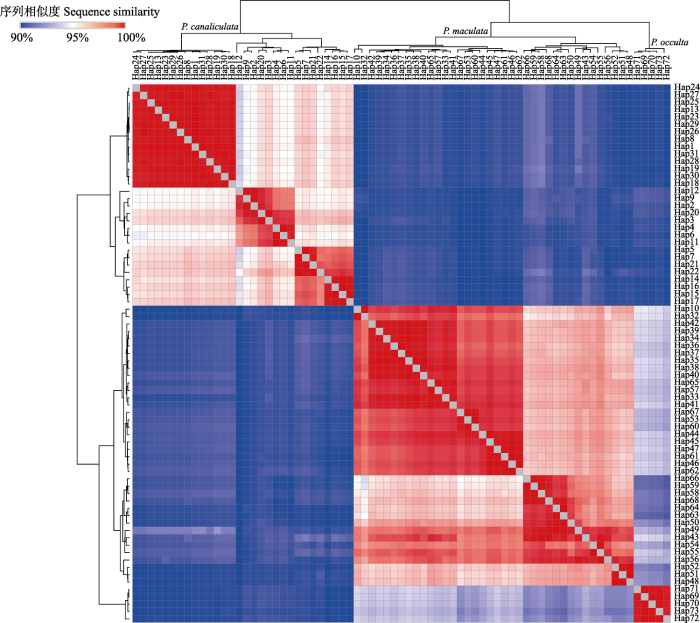
图2
基于K2P遗传距离的单倍型序列相似性热图。P. canaliculata: 小管福寿螺; P. maculata: 斑点福寿螺; P. occulta: 隐秘福寿螺。
Fig. 2
Heatmap of the sequence similarities based on K2P genetic distance of the haplotypes
图3
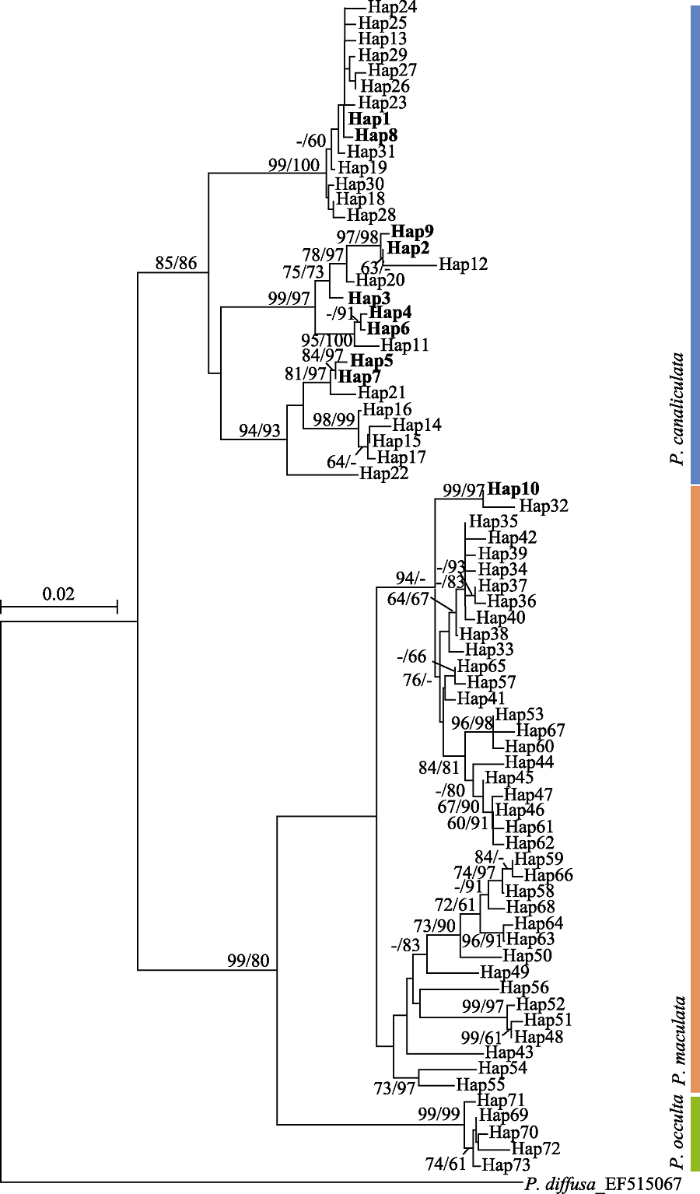
图3
长江下游福寿螺单倍型构建的系统发育树。系统发育分支节点为相邻连接法/贝叶斯法(NJ/BI)系统发育树的置信值, 仅显示> 60%的数值; 其中加粗显示的Hap1‒10为本研究所测COI序列生成的单倍型。
Fig. 3
The phylogenetic tree constructed by the haplotypes of apple snails from the lower reaches of the Yangtze River. The branch nodes of phylogenetic trees denote neighbor-joining/ Bayesian inference bootstrap supports. Only the values > 60% are displayed; Hap1-10 shown in bold are haplotypes generated from COI sequences from this study.
序列相似性和系统发育关系结果共同表明, 本研究种群共鉴定出两种福寿螺, Hap1-9为小管福寿螺, 包含234个样本, 占总样本数量的86.67%; Hap10为斑点福寿螺, 包含36个样品, 占比13.33%。
2.2 单倍型分布
小管福寿螺在所采集的11个地理种群中均有分布(Hap1-9), 上海种群(SHBS)仅分布有小管福寿螺, 包含Hap1和Hap7, 其中Hap7仅包含1条序列。斑点福寿螺(Hap10)仅分布在江苏地区长江以北的3个地理种群, 即扬州种群(JSYZ)、泰州种群(JSTZ)和宿迁种群(JSSQ); 仅泰州种群以斑点福寿螺为优势种(占76.67%), 其他种群均以小管福寿螺为优势种(表2)。
表2 本研究福寿螺种群的COI单倍型分布。地点编号见表1。
Table 2
| 单倍型 Haplotype | 序列数量 No. of sequences (%) | SHBS | WZLZ | WZMJB | KSZZ | CSSH | CSGL | ZJGFH | ZJGXSC | JSYZ | JSTZ | JSSQ | 物种 Species |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Hap1 | 126 (46.67) | 19 | 23 | 16 | 30 | 27 | - | - | - | - | 2 | 9 | 小管福寿螺Pomacea canaliculata |
| Hap2 | 15 (5.56) | - | - | - | - | - | - | 10 | - | - | 5 | - | 小管福寿螺 P. canaliculata |
| Hap3 | 21 (7.78) | - | - | - | - | - | 15 | 6 | - | - | - | - | 小管福寿螺 P. canaliculata |
| Hap4 | 28 (10.37) | - | - | - | - | - | - | - | 28 | - | - | - | 小管福寿螺 P. canaliculata |
| Hap5 | 22 (8.15) | - | 7 | 14 | - | 1 | - | - | - | - | - | - | 小管福寿螺 P. canaliculata |
| Hap6 | 18 (6.67) | - | - | - | - | - | - | - | - | 18 | - | - | 小管福寿螺 P. canaliculata |
| Hap7 | 1 (0.37) | 1 | - | - | - | - | - | - | - | - | 小管福寿螺 P. canaliculata | ||
| Hap8 | 2 (0.74) | - | - | - | - | 2 | - | - | - | - | 小管福寿螺 P. canaliculata | ||
| Hap9 | 1 (0.37) | - | - | - | - | - | - | 1 | - | - | - | - | 小管福寿螺 P. canaliculata |
| Hap10 | 36 (13.33) | - | - | - | - | - | - | - | - | 12 | 23 | 1 | 斑点福寿螺 P. maculata |
Hap1是小管福寿螺的优势单倍型, 包含126条序列, 占所测序列总数的46.67%, 分布于上海市(SHBS)、苏州市(WZLZ、WZMJB)、昆山市(KSZZ)、常熟市(CSSH)、泰州市(JSTZ)和宿迁市(JSSQ)等7个种群(表2)。Hap2-6分别包含15条、21条、28条、22条、18条序列; 其中, Hap2和Hap3各分布于2个种群, Hap4和Hap6分别仅在张家港市(ZJGXSC)和扬州市(JSYZ)分布, Hap5在苏州市吴中区(WZLZ、WZMJB)及常熟市(CSSH)发现(表2)。通过NCBI数据库的BLASTn比对发现, Hap8-9尚未在其他地区报道, 是本研究所检测到的特有单倍型, 分别分布于常熟种群(CSSH)和张家港种群(ZJGFH) (表2)。
2.3 小管福寿螺的种群遗传结构
本研究所构建的COI数据集共包含小管福寿螺30个单倍型(Hap1-9和Hap11-31, 附录3)。其中, Hap1是最大的单倍型, 共包含370条序列, 由阿根廷种群与各入侵地种群共享; Hap1是中国长江下游江苏和上海种群(126条)、香港种群(99条)以及日本种群(7条)的优势单倍型, 是已发表的中国大陆种群的第二大单倍型(129条)。Hap2-4由中国种群和日本种群共享: Hap2是已发表的中国大陆种群的最大单倍型(159条), 也是香港种群的第二大单倍型(32条), 但在中国江苏种群和日本种群分别仅检测到15条和1条序列; Hap3在中国江苏种群和日本种群分别包含多条序列, 但仅包含1条已发表的中国大陆种群序列(GenBank号KP310290); Hap4在张家港种群(ZJGXSC)、已发表的中国大陆种群和日本种群均包含多条序列, 但仅包含1条香港种群序列(附录3)。Hap5由中国的苏州市吴中区种群(WZLZ、WZMJB)、常熟市种群(CSSH), 以及日本种群共享; Hap6由江苏扬州种群、已发表中国大陆种群(浙江绍兴)和日本种群共享; Hap7是上海种群和阿根廷种群的共享单倍型(附录3, 表2)。
单倍型网络分析表明, 在95%的简约连接界限下小管福寿螺的单倍型共分成4个子网络A‒D, 本研究所测种群分布于单倍型网络A‒C, 网络D仅包含来自阿根廷种群的Hap22 (图4)。其中, 网络A‒B均除包含本研究测序种群外, 还包含中国大陆已发表种群、香港种群, 以及日本种群和阿根廷种群, 而网络C仅由本研究测序种群、日本种群和阿根廷种群共享(图4)。网络A包含14个单倍型, 以Hap1为中心, 其他单倍型以1-2步的距离分布于四周; 除Hap8和Hap13外, 其余均为阿根廷种群的单倍型(图4)。网络B包含8个单倍型, 仅Hap20来自阿根廷种群, 通过缺失单倍型与其他单倍型(Hap2、Hap3-4、Hap6、Hap9、Hap11-12)连接(图4)。网络C由中国的江苏、上海种群, 以及日本和阿根廷种群的单倍型共享, 除Hap5和Hap7外, 其余单倍型均来自阿根廷(表2, 图4)。
图4
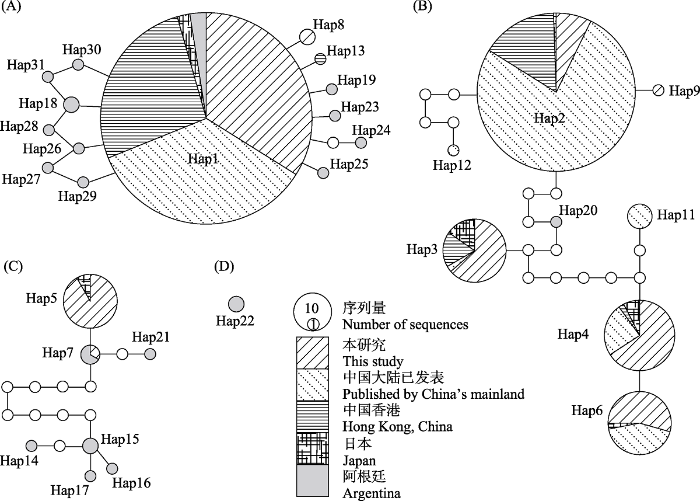
图4
基于TCS模型建立的小管福寿螺单倍型网络。95%简约连接界限下分成4个子网络(A‒D); 圆的大小与序列量成正比, 不同图案代表不同地理种群, 白色圆表示缺失单倍型。
Fig. 4
Haplotype network of Pomacea canaliculata based on TCS model. The haplotypes network of P. canaliculata splits into four sub-networks (A‒D) under 95% parsimony limit. The size of the circles represents the number of sequences. Pattern types represent different geographical populations, with the white circles indicating missing haplotypes.
2.4 斑点福寿螺的种群遗传结构
COI数据集所生成的单倍型中包含38个斑点福寿螺单倍型(Hap10和Hap32-68, 附录3)。在95%的简约连接界限下, 斑点福寿螺的单倍型网络分成7个子网络(A‒G); 其中, 除网络A外其他网络仅含有巴西种群的单倍型(图5)。网络A共包含23个单倍型, Hap10是最大的斑点福寿螺单倍型, 由中国的江苏种群、已发表的中国大陆种群、香港种群, 以及日本种群和巴西种群共享, 表明入侵来源于巴西。Hap32为已发表的中国大陆种群的单倍型, 与Hap10通过两个缺失单倍型连接; Hap33为香港种群单倍型, 通过1个缺失单倍型与阿根廷单倍型Hap35连接; Hap34由日本与阿根廷种群共享(附录3, 图5)。
图5
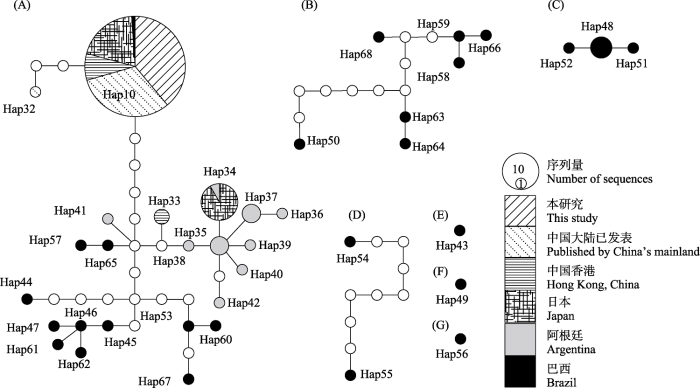
图5
基于TCS模型建立的斑点福寿螺单倍型网络。95%简约连接界限下分成7个子网络(A‒G); 圆的大小与序列量成正比, 不同图案代表不同地理种群, 白色圆表示缺失单倍型。
Fig. 5
Haplotype network of Pomacea maculata based on TCS model. The haplotypes network of P. maculata splits into seven sub-networks (A‒G) under 95% parsimony limit. The size of the circles represents the number of sequences. Pattern types represent different geographical populations, with the white circles indicating missing haplotypes.
2.5 种群遗传多样性
表3 本研究种群、已发表的中国大陆及香港种群的小管福寿螺和斑点福寿螺的遗传多样性
Table 3
| 物种 Species | 地理种群 Region | 序列数 No. of sequences | 单倍型数 No. of haplotypes | 单倍型多样性 Haplotype diversity (Hd) | 核苷酸多样性 Nucleotide diversity (π) | 核苷酸平均差异数 Average number of nucleotide difference (k) |
|---|---|---|---|---|---|---|
| 小管福寿螺 Pomacea canaliculata | 长江以南 South of the Yangtze River | 200 | 8 | 0.627 | 0.02546 | 14.744 |
| 长江以北 North of the Yangtze River | 34 | 3 | 0.611 | 0.02541 | 14.713 | |
| 中国大陆已发表 Published by China’s mainland | 319 | 7 | 0.587 | 0.02543 | 14.722 | |
| 香港 Hong Kong | 139 | 5 | 0.441 | 0.01956 | 11.324 | |
| 斑点福寿螺 P. maculata | 江苏 Jiangsu | 36 | 1 | 0.000 | 0.00000 | 0.000 |
| 中国大陆已发表 Published by China’s mainland | 30 | 2 | 0.067 | 0.00035 | 0.200 | |
| 香港 Hong Kong | 10 | 2 | 0.356 | 0.00430 | 2.489 |
AMOVA层次分析分组结果显示, 组群数设置为2‒7组时, 江苏和上海地区小管福寿螺均有长江以北种群与长江以南种群构成的组群, 分为7组时组间分化指数(FCT)最大为0.78503 (附录4)。在FCT = 0.77374时将所采集的小管福寿螺种群分成3大组群: 第一组群包括上海种群(SHBS)、太湖沿岸种群(WZLZ、WZMJB、KSZZ、CSSH)以及宿迁种群(JSSQ); 第二组群包括长江南岸的张家港(ZJGFH)、常熟古里(CSGL)以及泰州(JSTZ)种群; 第三组群包括张家港(ZJGXSC)和扬州(JSYZ)种群(图1, 附录4)。遗传变异组成表明, 组群间、组群内种群间以及种群内个体间变异分别占总变异的7.37%、7.24%和15.38%, 表明遗传变异主要来源于组群间(表4)。
表4 长江下游分布区小管福寿螺种群COI分子变异方差分析
Table 4
| 变异来源 Source of variation | 自由度 Degree of freedom | 平方和 Sum of squares | 方差组分 Variance components | 变异百分率 Percentage of variation (%) |
|---|---|---|---|---|
| 组群间 Among groups | 2 | 122.265 | 9.56034 | 7.37 |
| 组群内种群间 Among populations within groups | 8 | 165.622 | 0.89486 | 7.24 |
| 种群内个体间 Among individuals within populations | 223 | 423.865 | 1.90074 | 15.38 |
| 总变异 Total variation | 233 | 1,811.752 | 12.35594 | 100.00 |
2.6 种间杂交渐渗
基于特异引物多重PCR的方法针对核EF1α基因分型, 若扩增产物仅包含125 bp的片段则判断为小管福寿螺型(C型), 若仅包含151 bp的片段则判断为斑点福寿螺型(M型), 同时仅包含125 bp和151 bp的两条片段则判断为小管福寿螺和斑点福寿螺杂合型(B型) (图6) (Yang et al, 2020)。结合线粒体COI基因和核基因EF1α分析, 共检测到6种杂交型。在小管福寿螺中共检测到126个纯种(CC型), 占比53.85%; CB型和CM型分别占35.90%和10.26%。其中, 常熟古里种群(CSGL)和宿迁种群(JSSQ)的小管福寿螺均为纯种, 仅在上海种群(SHBS)、太湖沿岸种群(WZLZ、WZMJB、CSSH)和张家港种群(ZJGXSC) 4个种群检测到CM型。与小管福寿螺不同, 斑点福寿螺样本中杂种比例较高。其中, MB型和MC型分别检测到11个(30.56%)和23个(63.89%),仅在扬州种群(JSYZ)检测到2个纯种斑点福寿螺(MM型), 占比5.56%。
图6
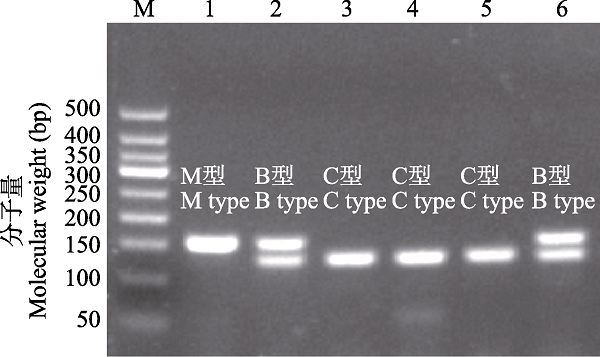
图6
基于核EF1α基因多重PCR扩增的长江下游福寿螺杂交渐渗型检测。M为50 bp DNA梯带; 1‒6为长江下游福寿螺样品, 分别为M型、B型、C型、C型、C型、B型。
Fig. 6
Introgressive hybridization of the apple snails in the lower reaches of the Yangtze River based on EF1α gene multiplex PCR method. M, 50 bp DNA ladder; 1‒6 represent for M type, B type, C type, C type, C type, and B type of samples of apple snails collected in the lower reaches of the Yangtze River.
3 讨论
福寿螺持续向冬季低温区扩散引发全球广泛关注。本研究针对长江下游南北分布区开展调研, 发现福寿螺向北已扩散至宿迁市泗洪县, 表明福寿螺在江苏大部分地区具有适生性。长江以北江苏地区有小管福寿螺和斑点福寿螺两个物种, 在长江以南的江苏种群和上海种群仅检测到小管福寿螺发生。已有研究显示我国斑点福寿螺主要发生在四川盆地、浙江、江苏和香港地区(Yang et al, 2018, 2022; 钱子衿等, 2021); 其中, 钱子衿等(2021)仅在长江以南的江苏苏州发现斑点福寿螺, 而本研究发现了长江以北江苏地区斑点福寿螺的分布, 进一步扩大了我们对该物种分布范围的认识。尽管隐秘福寿螺已在我国十余省份发生(Yang et al, 2018), 但我们未在长江下游地区检测到这一物种。小管福寿螺生成9个单倍型, 其中发现2个特有的单倍型(Hap8-9); 斑点福寿螺仅检测到1个单倍型。与原产地阿根廷和巴西福寿螺种群相比, 该地区的种群单倍型数量较少, 表明入侵种群经历了奠基者效应, 仅保留了少量祖先单倍型(Nei et al, 1975)。
已有报道表明, 小管福寿螺是亚洲许多入侵地的优势物种(Hayes et al, 2008; Yang et al, 2018)。本研究所采集的地点均有小管福寿螺分布, 但在长江以北地区的泰州市种群中斑点福寿螺为优势物种(占76.67%), 扬州市种群也具有较高的斑点福寿螺比例(60%)。这与所预期的斑点福寿螺低温耐受力低、难以向北扩散相悖(Matsukura et al, 2016)。例如, Matsukura等(2013)针对日本分布的福寿螺物种鉴定表明, 斑点福寿螺仅分布在南部地区, 在高纬低温的北部仅有小管福寿螺分布。本研究调研显示, 长江以北江苏地区福寿螺主要发生在河流、湖泊等深水生境, 在稻田等浅水湿地极少发生。这可能是由于冬季深水底部及淤泥中仍能保持较高的温度(Wang et al, 2019), 对福寿螺的存活起到保护作用有关。此外, 泰州种群主要在蟹田周边, 福寿螺的最初来源可能主要作为蟹饲料而人为引入的。由于斑点福寿螺与小管福寿螺具有相似的取食偏好性, 但斑点福寿螺具有更高的生长速率, 产卵多、生长速度快、成螺个体大等优势, 使其在养殖中更受青睐(Morrison & Hay, 2011; Hayes et al, 2012)。
单倍型组成与单倍型网络结构共同表明, 长江下游江苏和上海地区的小管福寿螺种群遗传结构与我国已发表的大陆和香港种群具有较大差异, 而与日本种群更相似。种群遗传结构表明, 长江下游江苏和上海地区的小管福寿螺可能从阿根廷多次入侵, 并发现一个新的单倍型网络(图4C), 表明该地区具有独立的入侵事件。多次入侵可以从原产地的基因库里获得更多的基因型, 是提升入侵种群遗传多样性的重要原因之一(Eyer et al, 2021)。这可能是导致长江下游江苏和上海分布区的小管福寿螺比我国大陆其他地区种群、香港种群等具有较高的种群遗传多样性的重要原因。种群遗传多样性对于外来生物在新生境中的环境适应性至关重要, 种群遗传多样性越高越有利于扩散(VanWallendael et al, 2021)。多次入侵及其所导致的较高的种群遗传多样性对福寿螺在长江下游江苏和上海地区的快速适应性进化和种群扩张具有重要促进作用。
江苏和上海地区长江南部和北部的小管福寿螺种群遗传多样性相似, 表明长江以南种群间、长江以北种群间均不具有明显的遗传分化; 然而, 长江下游江苏和上海地区的小管福寿螺种群形成跨长江分布的3个组群, 且组群间遗传变异是主要的分子变异来源。这一结果表明, 江苏和上海地区长江以北的小管福寿螺可能由长江以南种群扩散而来。中国四川盆地、浙江和长江以南江苏地区的斑点福寿螺来源于巴西, 而中国香港和日本的斑点福寿螺种群来源于巴西和阿根廷(Matsukura et al, 2008; Yang et al, 2018, 2022; 钱子衿等, 2021)。种群遗传结构分析表明, 中国长江以北江苏地区的斑点福寿螺与四川盆地和浙江种群的福寿螺一样, 均来源于巴西。福寿螺自主扩散能力有限, 主要通过引种等人类活动进行远距离扩散(Cowie et al, 2006)。本研究的斑点福寿螺种群仅形成1个单倍型, 这与中国大陆其他分布区的斑点福寿螺主单倍型相一致, 我们推测长江以北的斑点福寿螺可能由国内其他分布区随人类活动扩散而来。小管福寿螺与斑点福寿螺的种群共同揭示了长江以北江苏地区的福寿螺种群具有多元的入侵来源。
杂交是适应性进化的重要驱动因素(Kagawa & Takimoto, 2018)。基因渐渗是提升种群遗传多样性的重要机制之一(Freeland & Boag, 1999)。福寿螺种间渐渗杂交在原产地就已突破种间生殖障碍, 发生渐渗杂交(Glasheen et al, 2020), 并扩散到世界其他地区。其中, 乌拉圭和巴西的杂种比例约为30% (Glasheen et al, 2020)。针对核基因分型分析结果, 本研究中小管福寿螺和斑点福寿螺种群的杂交种比例均高于原产地, 表明渐渗杂交在入侵过程中持续发生, 可能在促进入侵扩散中发挥重要作用。此外, Yang等(2020)对中国30个种群的小管福寿螺和斑点福寿螺进行种间渐渗检测, 发现纯种小管福寿螺(CC型)比例为26.86%、线粒体为斑点福寿螺而核基因为小管福寿螺的MC型占比35.29%。与全国其他地区福寿螺种群相比, 本研究种群中CC型(53.85%)和MC型(63.89%)比例均较高。这可能是由于小管福寿螺耐寒性显著高于斑点福寿螺, 斑点福寿螺通过与小管福寿螺杂交提升了子代的耐低温水平(Matsukura et al, 2013, 2016)。此外, 当前仅在香港发现了1头纯种斑点福寿螺, 而在中国大陆、日本和韩国等地区尚未发现(Matsukura et al, 2016; Yang et al, 2020, 2022)。本研究在江苏省扬州市种群检测到2头纯种斑点福寿螺样本, 这是否与上述深水生境的温度保护作用或渐渗杂交对福寿螺低温适应性的影响机制有关, 仍有待深入研究。
综上所述, 本研究探明了长江下游江苏和上海地区的福寿螺种类及分布, 明确了该地区长江南北福寿螺的种群遗传结构, 揭示了长江以北江苏地区的福寿螺具有多元的入侵来源, 并首次发现了小管福寿螺在中国的一个新的入侵历史事件。多元入侵来源、种间渐渗杂交等因素有利于提升福寿螺的种群遗传多样性, 进而对其快速适应环境及扩张种群提供了有利的遗传基础。全球气候变暖以及频繁的水运、灌溉、引种等人类活动, 为福寿螺持续向北扩散提供了有利条件。鉴于福寿螺有进一步向我国北方扩散的风险, 有必要严格检疫、加强监测, 深入开展福寿螺低温可塑性内在机制研究, 同时加强植物保护、水生生态、疾控中心、水利及交通运输等多部门协同, 对福寿螺的扩散范围和发生程度进行有效控制。
致谢
感谢中国计量大学的李迦南、赵星星、高跃, 昆山市耕保植保植检站的周斌, 常熟市植保植检站的黄亚川, 常熟市尚湖镇农技推广服务中心的顾立丹在样品采集中给予的支持与帮助。
附录 Supplementary Material
附录1 本研究采集的福寿螺外壳形态
Appendix 1 Shell morphology of apple snails collected from this study
附录2 从GenBank中下载的福寿螺COI序列及参考文献
Appendix 2 The COI sequences of apple snails retrieved from GenBank and their references
附录3 本研究及已发表的中国大陆、香港以及日本、阿根廷、巴西福寿螺种群的COI单倍型分布及序列信息
Appendix 3 The COI haplotype distribution and sequence information of apple snails from this study and the populations published by China’s mainland, Hong Kong, and Japan, Argentina, Brazil
附录4 基于AMOVA层次分析的本研究小管福寿螺种群的分组
Appendix 4 Grouping of the Pomacea canaliculata populations from this study based on AMOVA hierarchical analysis
参考文献
The golden apple snail Pomacea canaliculata: A review on invasion, dispersion and control
DOI:10.1564/v27_aug_03 URL [本文引用: 1]
TCS: A computer program to estimate gene genealogies
DOI:10.1046/j.1365-294x.2000.01020.x PMID:11050560 [本文引用: 1]
What are apple snails? Confused taxonomy and some preliminary resolution
In: Global Advances in Ecology and Management of Golden Apple Snails (eds Joshi RC, Sebastian LS), pp. 3-23. Philippine Rice Research Institute, Munñoz.
Analysis of molecular variance inferred from metric distances among DNA haplotypes: Application to human mitochondrial DNA restriction data
DOI:10.1093/genetics/131.2.479
PMID:1644282
[本文引用: 1]

We present here a framework for the study of molecular variation within a single species. Information on DNA haplotype divergence is incorporated into an analysis of variance format, derived from a matrix of squared-distances among all pairs of haplotypes. This analysis of molecular variance (AMOVA) produces estimates of variance components and F-statistic analogs, designated here as phi-statistics, reflecting the correlation of haplotypic diversity at different levels of hierarchical subdivision. The method is flexible enough to accommodate several alternative input matrices, corresponding to different types of molecular data, as well as different types of evolutionary assumptions, without modifying the basic structure of the analysis. The significance of the variance components and phi-statistics is tested using a permutational approach, eliminating the normality assumption that is conventional for analysis of variance but inappropriate for molecular data. Application of AMOVA to human mitochondrial DNA haplotype data shows that population subdivisions are better resolved when some measure of molecular differences among haplotypes is introduced into the analysis. At the intraspecific level, however, the additional information provided by knowing the exact phylogenetic relations among haplotypes or by a nonlinear translation of restriction-site change into nucleotide diversity does not significantly modify the inferred population genetic structure. Monte Carlo studies show that site sampling does not fundamentally affect the significance of the molecular variance components. The AMOVA treatment is easily extended in several different directions and it constitutes a coherent and flexible framework for the statistical analysis of molecular data.
Extensive human-mediated jump dispersal within and across the native and introduced ranges of the invasive termite Reticulitermes flavipes
DOI:10.1111/mec.v30.16 URL [本文引用: 1]
DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates
We describe "universal" DNA primers for polymerase chain reaction (PCR) amplification of a 710-bp fragment of the mitochondrial cytochrome c oxidase subunit I gene (COI) from 11 invertebrate phyla: Echinodermata, Mollusca, Annelida, Pogonophora, Arthropoda, Nemertinea, Echiura, Sipuncula, Platyhelminthes, Tardigrada, and Coelenterata, as well as the putative phylum Vestimentifera. Preliminary comparisons revealed that these COI primers generate informative sequences for phylogenetic analyses at the species and higher taxonomic levels.
The mitochondrial and nuclear genetic homogeneity of the phenotypically diverse Darwin’s ground finches
DOI:10.1111/j.1558-5646.1999.tb05418.x
PMID:28565556
[本文引用: 1]
The most extensively studied group of Darwin's finches is the genus Geospiza, the ground finches, and yet little is known about the evolutionary history and genetic relationships of these birds. Studies using either allozyme or morphological data have been unable to resolve relationships between the six species and numerous populations of ground finches. In this paper we report the results of a study using mitochondrial control region and nuclear internal transcribed spacer (ITS) 1 sequence data. The differentiation of the ground finch species based on morphological data is not reflected in either mitochondrial or nuclear DNA sequence phylogenies. Furthermore, there is little concordance between the mitochondrial haplotypes and ITS alleles found within individuals. We suggest that the absence of species-specific lineages can be attributed to ongoing hybridization involving all six species of Geospiza. There are no long term selective pressures against hybridization within this genus, and therefore a genetically homogenous genus may be maintained indefinitely. Hybridization has apparently played a role in the adaptive radiation of Darwin's finches.© 1999 The Society for the Study of Evolution.
First evidence of introgressive hybridization of apple snails (Pomacea spp.) in their native range
DOI:10.1093/mollus/eyz035
PMID:32362703
[本文引用: 4]
Genetic variation facilitates both natural range expansions and anthropogenic invasions. Contrary to expectations, hybridization does not always impact negatively on biodiversity. Increasing evidence indicates advantageous roles for introgressive hybridization in maintaining standing genetic variation. Hypothesizing that hybridization may contribute to the evolutionary and invasive success of a diverse group of freshwater snails (Ampullariidae, commonly known as apple snails), we estimated the frequency of hybridization between two globally invasive species of, (Lamarck, 1822) and Perry, 1810, in their native range. While previous work in Asia has uncovered the occurrence of extensive hybridization, we provide the first phylogenetic evidence of a high degree of hybridization (30%) between these species in Uruguay and Brazil. Hybrids carried both heterozygous and homozygous combinations of elongation factor 1-α (EF1α) nuclear alleles in both mating directions, indicating that hybridization has occurred over multiple generations and likely preceded introductions outside the native range. Among the five sites in Brazil previously documented as containing only, one far northern population (Careiro Castanho), which is thousands of kilometres from the northern range of, unexpectedly contained hybrids. This may be the result of human-facilitated introductions. Together with recent work from Asia, our investigations in the native range of apple snails support a reframing of historical perspectives of hybridization as a driver of extinction and diversity loss towards a modern paradigm where hybridization may promote diversification and contribute to the survival of evolutionary lineages such as molluscs.© The Author(s) 2020. Published by Oxford University Press on behalf of The Malacological Society of London, all rights reserved. For Permissions, please email: journals.permissions@oup.com.
Insights from an integrated view of the biology of apple snails (Caenogastropoda: Ampullariidae)
DOI:10.4002/040.058.0209 URL [本文引用: 1]
A global phylogeny of apple snails: Gondwanan origin, generic relationships, and the influence of outgroup choice (Caenogastropoda: Ampullariidae)
DOI:10.1111/j.1095-8312.2009.01246.x URL [本文引用: 1]
Comparing apples with apples: Clarifying the identities of two highly invasive Neotropical Ampullariidae (Caenogas- tropoda)
DOI:10.1111/zoj.2012.166.issue-4 URL [本文引用: 3]
Out of South America: Multiple origins of non-native apple snails in Asia
DOI:10.1111/j.1472-4642.2008.00483.x URL [本文引用: 4]
Investigation on the distribution of apple snail (Pomacea canaliculata) in the lower reaches of Yangtze River in China
中国长江下游地区福寿螺分布现状考察
Hybridization can promote adaptive radiation by means of transgressive segregation
DOI:10.1111/ele.12891
PMID:29243294
[本文引用: 2]
Understanding the mechanisms of rapid adaptive radiation has been a central problem of evolutionary ecology. Recently, there is a growing recognition that hybridization between different evolutionary lineages can facilitate adaptive radiation by creating novel phenotypes. Yet, theoretical plausibility of this hypothesis remains unclear because, for example, hybridization can negate pre-existing species richness. Here, we theoretically investigate whether and under what conditions hybridization promotes ecological speciation and adaptive radiation using an individual-based model to simulate genome evolution following hybridization between two allopatrically evolved lineages. The model demonstrated that transgressive segregation through hybridization can facilitate adaptive radiation, most powerfully when novel vacant ecological niches are highly dissimilar, phenotypic effect size of mutations is small and there is moderate genetic differentiation between parental lineages. These results provide a theoretical basis for the effect of hybridization facilitating adaptive radiation.© 2017 The Authors. Ecology Letters published by CNRS and John Wiley & Sons Ltd.
Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data
DOI:10.1093/bioinformatics/bts199
PMID:22543367
[本文引用: 2]
The two main functions of bioinformatics are the organization and analysis of biological data using computational resources. Geneious Basic has been designed to be an easy-to-use and flexible desktop software application framework for the organization and analysis of biological data, with a focus on molecular sequences and related data types. It integrates numerous industry-standard discovery analysis tools, with interactive visualizations to generate publication-ready images. One key contribution to researchers in the life sciences is the Geneious public application programming interface (API) that affords the ability to leverage the existing framework of the Geneious Basic software platform for virtually unlimited extension and customization. The result is an increase in the speed and quality of development of computation tools for the life sciences, due to the functionality and graphical user interface available to the developer through the public API. Geneious Basic represents an ideal platform for the bioinformatics community to leverage existing components and to integrate their own specific requirements for the discovery, analysis and visualization of biological data.Binaries and public API freely available for download at http://www.geneious.com/basic, implemented in Java and supported on Linux, Apple OSX and MS Windows. The software is also available from the Bio-Linux package repository at http://nebc.nerc.ac.uk/news/geneiousonbl.
Using ensemble forecasting to examine how climate change promotes worldwide invasion of the golden apple snail (Pomacea canaliculata)
DOI:10.1007/s10661-017-6124-y
PMID:28726175
[本文引用: 1]
The golden apple snail, Pomacea canaliculata, is one of the world's 100 most notorious invasive alien species. Knowledge about the critical climate variables that limit the global distribution range of the snail, as well as predictions of future species distributions under climate change, is very helpful for management of snail. In this study, the climatically suitable habitats for this kind of snail under current climate conditions were modeled by biomod2 and projected to eight future climate scenarios (2 time periods [2050s, 2080s] × 2 Representative Concentration Pathways [RCPs; RCP2.6, RCP8.5] × 2 atmospheric General Circulation Models [GCMs; Canadian Centre for Climate Modelling and Analysis (CCCMA), Commonwealth Scientific and Industrial Research Organisation (CSIRO)]). The results suggest that the lowest temperature of coldest month is the critical climate variable to restrict the global distribution range of P. canaliculata. It is predicted that the climatically suitable habitats for P. canaliculata will increase by an average of 3.3% in 2050s and 3.8% in 2080s for the RCP2.6 scenario, while they increase by an average of 8.7% in 2050s and 10.3% in 2080s for the RCP8.5 scenario. In general, climate change in the future may promote the global invasion of the invasive species. Therefore, it is necessary to take proactive measures to monitor and preclude the invasion of this species.
Population genetic structure of Gonionemus vertens based on the mitochondrial COI sequence
DOI:10.17520/biods.2018044 URL [本文引用: 1]
基于线粒体COI基因分析钩手水母的群体遗传结构
DOI:10.17520/biods.2018044
[本文引用: 1]
钩手水母(Gonionemus vertens)为大西洋和太平洋广布种, 是我国习见的有毒水母种类之一。本文对采自黄渤海海域4个地理群体的104个钩手水母线粒体COI基因序列进行扩增, 并结合GenBank上其他182个钩手水母同源序列进行序列变异分析。在286个基因序列中共检测出52个多态位点, 定义了14种单倍型。总群体的单倍型多样性和核苷酸多样性分别为0.743 ± 0.012和1.046% ± 0.097%, 与其他几种大型水母相比, 钩手水母总群体的遗传多样性处于较高水平。AMOVA结果显示, 60.17%的分子变异源于群组间, 13.37%的分子变异源于群体内, 26.46%的分子变异源于组内群体间, 群组间、群体内和组内群体间的遗传分化均极显著。F<sub>st</sub>值统计检验表明, 中国厦门群体与乐亭、东营、烟台、大连群体间存在显著的遗传分化, 大连与东营、烟台群体间也存在显著的遗传分化。系统分析结果显示, 钩手水母群体间存在2个明显的单倍型谱系分支。不同的钩手水母地理群体间具有复杂的遗传模式, 钩手水母复杂的生活史、扩散能力、地理隔离和海流分布可能是影响钩手水母遗传结构的重要因素。
Investigation on northernmost distribution areas of apple snail (Pomacea canaliculata) in upper reaches of Yangtze River
长江上游地区福寿螺北缘分布地区调查
Correlation between shell-body mass ratio and hydrostatic settling characteristics of mollusc species
贝类壳-体质量比和静水沉降特性的相关性
The emergence of angiostrongyliasis in the People’s Republic of China: The interplay between invasive snails, climate change and transmission dynamics
DOI:10.1111/fwb.2011.56.issue-4 URL [本文引用: 1]
Cold tolerance of invasive freshwater snails, Pomacea canaliculata, P. maculata, and their hybrids helps explain their different distributions
DOI:10.1111/fwb.12681 URL [本文引用: 4]
Genetic exchange between two freshwater apple snails, Pomacea canaliculata and Pomacea maculata invading East and Southeast Asia
DOI:10.1007/s10530-013-0431-1 URL [本文引用: 4]
Genetic divergence of the genus Pomacea (Gastropoda: Ampullariidae) distributed in Japan, and a simple molecular method to distinguish P. canaliculata and P. insularum
DOI:10.1303/aez.2008.535 URL [本文引用: 2]
Feeding and growth of native, invasive and non-invasive alien apple snails (Ampullariidae) in the United States: Invasives eat more and grow more
DOI:10.1007/s10530-010-9881-x URL [本文引用: 1]
The bottleneck effect and genetic variability in populations
DOI:10.1111/j.1558-5646.1975.tb00807.x PMID:28563291 [本文引用: 1]
Genetic diversity of invasive Pomacea snails in Suzhou City
苏州市入侵福寿螺的遗传多样性
The identity, distribution, and impacts of non-native apple snails in the continental United States
Since the mid 1990s populations of non-native apple snails (Ampullariidae) have been discovered with increasing frequency in the continental United States. Given the dramatic effects that introduced apple snails have had on both natural habitats and agricultural areas in Southeast Asia, their introduction to the mainland U.S. is cause for concern. We combine phylogenetic analyses of mtDNA sequences with examination of introduced populations and museum collections to clarify the identities, introduced distributions, geographical origins, and introduction histories of apple snails.Based on sampling to date, we conclude there are five species of non-native apple snails in the continental U.S. Most significantly, we recognize three species within what has been called the channeled apple snail: Pomacea canaliculata (California and Arizona), Pomacea insularum, (Florida, Texas, and Georgia) and Pomacea haustrum (Florida). The first established populations of P. haustrum were discovered in the late 1970s in Palm Beach County Florida, and have not spread appreciably in 30 years. In contrast, populations of P. insularum were established in Texas by 1989, in Florida by the mid to late 1990s, and in Georgia by 2005, and this species continues to spread rapidly. Most introduced P. insularum haplotypes are a close match to haplotypes from the Río Uruguay near Buenos Aires, indicating cold tolerance, with the potential to spread from Florida, Georgia, and Texas through Louisiana, Alabama, Mississippi, and South Carolina. Pomacea canaliculata populations were first discovered in California in 1997. Haplotypes of introduced P. canaliculata match native-range haplotypes from near Buenos Aires, Argentina, also indicating cold tolerance and the potential to establish farther north.The term "channeled apple snail" is descriptive of a morphology found in many apple snail species. It does not identify a single species or a monophyletic group. Clarifying species identifications permits a more accurate assessment of introduction histories and distributions, and provides a very different picture of the tempo and pattern of invasions than was inferred when the three species with channeled sutures were considered one. Matching introduced and native-range haplotypes suggests the potential for range expansion, with implications for native aquatic ecosystems and species, agriculture, and human health.
MrBayes 3: Bayesian phylogenetic inference under mixed models
DOI:10.1093/bioinformatics/btg180
PMID:12912839
[本文引用: 1]
MrBayes 3 performs Bayesian phylogenetic analysis combining information from different data partitions or subsets evolving under different stochastic evolutionary models. This allows the user to analyze heterogeneous data sets consisting of different data types-e.g. morphological, nucleotide, and protein-and to explore a wide variety of structured models mixing partition-unique and shared parameters. The program employs MPI to parallelize Metropolis coupling on Macintosh or UNIX clusters.
DnaSP 6: DNA sequence polymorphism analysis of large data sets
DOI:10.1093/molbev/msx248
PMID:29029172
[本文引用: 2]
We present version 6 of the DNA Sequence Polymorphism (DnaSP) software, a new version of the popular tool for performing exhaustive population genetic analyses on multiple sequence alignments. This major upgrade incorporates novel functionalities to analyze large data sets, such as those generated by high-throughput sequencing technologies. Among other features, DnaSP 6 implements: 1) modules for reading and analyzing data from genomic partitioning methods, such as RADseq or hybrid enrichment approaches, 2) faster methods scalable for high-throughput sequencing data, and 3) summary statistics for the analysis of multi-locus population genetics data. Furthermore, DnaSP 6 includes novel modules to perform single- and multi-locus coalescent simulations under a wide range of demographic scenarios. The DnaSP 6 program, with extensive documentation, is freely available at http://www.ub.edu/dnasp.© The Author 2017. Published by Oxford University Press on behalf of the Society for Molecular Biology and Evolution. All rights reserved. For permissions, please e-mail: journals.permissions@oup.com.
Patterns of population genomic diversity in the invasive Japanese knotweed species complex
DOI:10.1002/ajb2.1653
PMID:33942283
[本文引用: 1]
Invasive species are expected to undergo a reduction in genetic diversity due to founder effects, which should limit their ability to adapt to new habitats. Still, many invasive species achieve widespread distributions and dense populations. This paradox of invasions could potentially be overcome through multiple introductions or hybridization, both of which increase genetic diversity. We conducted a population genomics study of Japanese knotweed (Reynoutria japonica), which is a polyploid, clonally reproducing invasive species that has been notoriously successful worldwide despite supposedly low genetic diversity.We used genotyping by sequencing to collect 12,912 SNP markers from 88 samples collected at 38 locations across North America for the species complex. We used alignment-free k-mer hashing analysis in addition to traditional population genetic analyses to account for the challenges of genotyping polyploids.Genotypes conformed to three genetic clusters, likely representing Japanese knotweed, giant knotweed, and hybrid bohemian knotweed. We found that, contrary to previous findings, the Japanese knotweed cluster had substantial genetic diversity, though it had no apparent genetic structure across the landscape. In contrast, giant knotweed and hybrids showed distinct population groups. We did not find evidence of isolation by distance in the species complex, likely reflecting the stochastic introduction history of this species complex.The results indicate that clonal invasive species can show substantial genetic diversity and can be successful at colonizing a variety of habitats without showing evidence of local adaptation or genetic structure.© 2021 Botanical Society of America.
Estimating ecological flows for fish overwintering in plain rivers using a method based on water temperature and critical water depth
Prediction of potential geographical distribution area of golden apple snail (Pomacea canaliculata) in China
福寿螺在中国的潜在地理分布区预测
Introgressive hybridization between two non-native apple snails in China: Widespread hybridization and homogenization in egg morphology
DOI:10.1002/ps.v76.12 URL [本文引用: 5]
Molecular analyses revealed three morphologically similar species of non-native apple snails and their patterns of distribution in freshwater wetlands of Hong Kong
DOI:10.1111/ddi.v28.1 URL [本文引用: 6]
Invisible apple snail invasions: Importance of continued vigilance and rigorous taxonomic assessments
DOI:10.1002/ps.5241
PMID:30324686
[本文引用: 3]
Due to the similarities of overall shell morphology among apple snail species and considerable variability within species, substantial taxonomic confusion has plagued the accurate identification of Pomacea species. Many invasive apple snails have been mistakenly identified as P. canaliculata since their introduction to Asia around 1980. In 2008, three other introduced species in addition to P. canaliculata were recognized. In 2013, a fifth, previously unrecognized lineage was reported from China, indicating that despite the taxonomic clarity brought by previous work, continued surveys and taxonomic research are necessary to prevent additional introductions and continued spread, as well as to develop effective management strategies.Phylogenetic analysis of mitochondrial COI sequences confirmed the presence of a widespread unidentified Pomacea lineage in China. All sequences from samples of this newly documented lineage were recovered in a monophyletic clade delineated from closely related species; however, different DNA barcoding methods yielded inconsistent species boundaries. Additionally, nuclear EF1α sequences indicated incomplete lineage sorting or recent hybridization of the unidentified lineage with the other two established species.Barcoding is a valuable tool for species discovery, and a powerful approach for delineating introduced species. However, determining the identity of the newly discovered invasive lineage in China will require an integrated taxonomic approach incorporating individuals from the native range, and examination of natural history collections at museums around the world. To manage and prevent additional spread of already established species, and to stop the introduction of new taxa, continued monitoring and rigorous taxonomic assessments must be undertaken. © 2018 Society of Chemical Industry.© 2018 Society of Chemical Industry.
Distribution and the origin of invasive apple snails, Pomacea canaliculata and P. maculata (Gastropoda: Ampullariidae) in China
DOI:10.1038/s41598-017-19000-7
Species of Pomacea, commonly known as apple snails, are native to South America, and have become widely distributed agricultural and environmental pests in southern China since their introduction in the 1980s. However, only since 2010 have researchers recognized that at least two species, P. canaliculata and P. maculata, are present in China. Although impacts of apple snails have been extensively documented, confusion still persists regarding current distributions and origin of the species in China. To resolve this confusion, we used phylogenetic and phylogeographic methods to analyze 1464 mitochondrial COI sequences, including 349 new sequences from samples collected in southern China and 1115 publicly available sequences from snails collected in the native and introduced ranges. Pomacea canaliculata was found at all sampled localities, while P. maculata was found at only five sampled localities in the Sichuan basin and Zhejiang province. Our data indicate that Chinese populations of P. canaliculata share an Argentinian origin, consistent with multiple introductions of this species elsewhere in Asia. In addition, just a single lineage ofP. maculata is established in China, which shares with populations in Brazil.
Molecular identification of invasive golden apple snails in Zhejiang Province based on DNA barcoding
DOI:10.17520/biods.2015260
[本文引用: 1]
Golden apple snails seriously damage crops and aquatic ecosystem in China. Two invasive apple snail species, Pomacea canaliculata and P. maculata, have been reported in China since 2010. Only the distribution of P. canaliculata was reported in Zhejiang Province. It is difficult to distinguish the two species due to their close morphological characteristics and high diversity of shell morphology, which are influenced by both environmental factors and food types. We collected samples from seven localities from Zhejiang Province and sequenced 101 individuals of mitochondrial COI fragments of the DNA barcode region. We also downloaded 55 sequences of five species of the P. canaliculata group, which included all public sequences of P. lineata, P. dolioides, and P. paludosa, and South American sample sequences of P. canaliculata and P. maculata. Analyses including similarity alignments, DNA barcoding gaps, and phylogenetic relationships, revealed that COI sequences were effective to distinguish apple snail species. We detected P. canaliculata and P. maculata distributed in the Jianggan region of Hangzhou, while only P. canaliculata was distributed in the Putuo region of Zhoushan, the Shanyu and Xinchang regions of Shaoxing, the Ouhai region of Wenzhou and the Xihu regoin of Hangzhou. P. canaliculata revealed a much wider distribution range. P. canaliculata and P. maculata were collapsed into 4 haplotypes and 2 haplotypes, respectively. There were 1 to 3 haplotypes in each locality, which indicated a low genetic diversity. The phylogenetic analyses deduced that P. canaliculata and P. maculata were probably introduced from Argentina and Brazil, respectively.
基于DNA条形码技术对浙江省外来入侵福寿螺进行分子鉴定
DOI:10.17520/biods.2015260
[本文引用: 1]
外来入侵福寿螺对我国农业生产和水生生态系统平衡等造成严重危害。2010年, 种类鉴定研究首次揭示我国外来入侵福寿螺包括Pomacea canaliculata和P. maculata两个种, 而浙江省仅见P. canaliculata一种报道。P. canaliculata和P. maculata种间形态近似, 且受环境、食物源等因素影响, 同种内外壳形态特征多样, 因而基于形态特征进行种类的准确鉴定极为困难。本研究在采集浙江省7个区县的福寿螺样本的基础上, 利用DNA条形码技术扩增了101个不同个体的COI序列, 并从BOLD数据库下载了“P. canaliculata种团”的5个近缘种的55条COI序列用于分析, 其中包括P. lineata, P. dolioides和P. paludosa所有已发表序列, 以及P. canaliculata和P. maculata的南美洲样品的序列等。序列相似度比对、DNA条形码间隙和系统发育树等分析表明, COI序列可以实现近缘福寿螺的有效鉴别。待测的浙江省福寿螺样品中, 杭州江干区检测到P. canaliculata和P. maculata两种, 而舟山普陀区、绍兴上虞区和新昌县、温州瓯海区及杭州西湖区仅检测到P. canaliculata, 表明P. canaliculata在浙江省具有更广的分布范围。P. canaliculata和P. maculata分别形成4种和2种单倍型, 各区县样点分别包含1-3种单倍型, 浙江省各发生地呈现较低的遗传多样性。依据系统发育关系推测, 浙江省分布的P. canaliculata和P. maculata分别可能来源于阿根廷和巴西。
A new species of apple snail in the genus Pomacea (Gastropoda: Caenogastropoda: Ampullarii- dae)
Historical invasion, expansion process and harm investigation of Pomacea canaliculata in China
福寿螺在中国的入侵历史、扩散规律和危害的调查分析
Predicting current potential distribution and the range dynamics of Pomacea canaliculata in China under global climate change
DOI:10.3390/biology11010110
URL
[本文引用: 1]
Pomacea canaliculata is one of the 100 worst invasive alien species in the world, which has significant effects and harm to native species, ecological environment, human health, and social economy. Climate change is one of the major causes of species range shifts. With recent climate change, the distribution of P. canaliculata has shifted northward. Understanding the potential distribution under current and future climate conditions will aid in the management of the risk of its invasion and spread. Here, we used species distribution modeling (SDM) methods to predict the potential distribution of P. canaliculata in China, and the jackknife test was used to assess the importance of environmental variables for modeling. Our study found that precipitation of the warmest quarter and maximum temperature in the coldest months played important roles in the distribution of P. canaliculata. With global warming, there will be a trend of expansion and northward movement in the future. This study could provide recommendations for the management and prevention of snail invasion and expansion.
Tolerance to low temperature and desiccation in two invasive apple snails, Pomacea canaliculata and P. maculata (Caenogastropoda: Ampullariidae), collected in their original distribution area (northern and central Argentina)
DOI:10.1093/mollus/eyt042 URL [本文引用: 1]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}