



土壤生态系统高度复杂, 具有庞大的生物多样性(Decaëns, 2010)。以往, 专家和学者们对生物多样性的研究主要集中在水生和陆生(地上部分)生物; 近年来, 有关土壤生物的多样性研究才越来越受关注和重视(Bardgett & van der Putten, 2014; Phillips et al, 2020; Thakur et al, 2020)。土壤动物是土壤生态系统的重要组成部分, 在土壤的形成、改善土壤环境、促进土壤物质循环和能量流动等方面扮演着重要的角色(张志丹等, 2012; 战丽莉, 2013), 是陆地生态系统中种类和数量仅次于土壤微生物的类群(潘开文等, 2016)。不同土壤动物之间体型大小有很大的差异, 根据体长可分为3类: 大型(体长大于2 mm)、中型(体长在0.2 mm和2 mm之间)和小型(体长小于0.2 mm)土壤动物(Lavelle et al, 2006)。
近年来兴起的高通量测序技术(high throughput sequencing, HTS)为多样性调查和监测提供了可靠、高效的鉴定(分类预测)新思路(Taberlet et al, 2012)。随着分子测序技术的发展, DNA宏条形码(metabarcoding)技术开始广泛应用于多样性调查和监测的分类预测环节(Bista et al, 2018; Arribas et al, 2021)。宏条形码技术通过对从环境样品(包括水、土壤、空气等)或生物混合样品中收集的混合DNA进行靶标标记(常称为“条形码”)扩增, 并利用高通量测序技术得到大量可操作分类单元(operational taxonomic units, OTUs), 最后将得到的OTUs与分子数据库中的参照序列进行比对以实现分子鉴定(Ji et al, 2013; Bohmann et al, 2014)。与形态学方法相对应, 像DNA宏条形码技术这样, 通过借助于测序技术得到生物DNA等遗传物质来实现其分子层面的分类鉴定的预测方法称为分子分类预测。
宏条形码技术最初应用于环境微生物, 环境中很多微生物不能直接分离培养, 只能通过混合样品来获取遗传信息(刘莉扬等, 2013)。近年来, 宏条形码技术开始扩展到动物研究领域, 例如鱼类(Stat et al, 2017; Sales et al, 2021)和两栖动物(Bálint et al, 2018)等。然而, 它在土壤动物中的应用还未得到普及, 该类群的条形码数据库也极度匮乏。尽管如此, 土壤生物多样性研究者们不断做出尝试以推进宏条形码技术在土壤动物中的应用。Oliverio等(2018)曾尝试使用宏条形码技术在目及科阶元对土壤动物进行分类预测, 结果表明分子分类预测技术能准确将土壤动物鉴定到高阶元; Arribas等(2021)将单倍型(amplicon sequence variants, ASVs)水平的宏条形码应用于土壤动物, 结果与基于阈值的OTU多样性趋势一致。然而, 基于宏条形码技术的分子分类预测对土壤动物在低阶元水平(科以下)的表现依旧缺乏定量研究。
准确而高效的分子分类预测对生物多样性调查和监测至关重要(Bazinet & Cummings, 2012), 分子数据库的完备性和分子分类预测软件的选择决定了分类预测的准确性和效率。分子数据库的完备程度严重受制于分类工作者的进展等限制(时雷雷和傅声雷, 2014), 但分子分类预测工具和算法的迅猛发展使得分子分类在数据库受限的情况下仍有进一步提升的空间。近年来出现了各类分子分类预测软件, 主要基于两种算法。一种是传统的基于相似度的算法, 在覆盖率足够高的前提下, 通过将目标序列与数据库的序列进行比对, 得到与目标序列相似度最高的一条或多条参考序列的分类信息作为目标序列的分类预测参考。我们常用的BLAST (Altschul et al, 1990)就是基于这种算法实现分类预测的。另一种算法基于系统发育位置(phylogenetic placement, PP), 将目标序列放置到参考序列构建的系统发育树的各个分支上并计算目标序列在各个分支上的可能性, 最终计算出概率最高的分支从而预测出目标序列的分类信息(Berger & Stamatakis, 2011), 如EPA (RAxML) (Berger et al, 2011)等。
以往的比较和评估分子分类预测软件的研究主要基于相对完整的参考数据库(Brandt et al, 2021; Hleap et al, 2021; Mathon et al, 2021), 但不幸的是, 高度复杂多样的土壤动物的条形码数据库极度不完整, 这导致了在现实研究中将混合样品或环境样品中的土壤动物预测到物种水平十分困难。尽管如此, 我们依旧可以尝试在科级水平甚至属级水平进行分类预测。基于宏条形码技术的分子分类预测技术在动物应用中主要使用3种条形码: COI、16S和18S。其中, COI (线粒体细胞色素c氧化酶亚基I基因, cytochrome c oxidase I)的使用最为广泛, 因为动物的COI参考序列较其他条形码标记拥有更高的物种覆盖度(Hebert et al, 2003; Braukmann et al, 2019)。然而, Deagle等(2014)认为, COI的扩增引物保守性过低, 使用它来恢复目标类群可能会影响宏条形码分类预测技术的准确性。因此, 部分研究开始尝试用更为保守的16S (Elbrecht et al, 2016)和18S (Yang, 2013)作为分子标记。值得关注的是, 在Yang (2013)的研究中, 18S扩增出了比COI更为广泛的土壤动物类群。因此, 本研究同时采用了这3种条形码以表征不同程度的保守性和分子标记的差异。
本研究把多个分子分类预测软件的代码集成于一个可以统一执行的脚本, 可以高效快速地输出所有目标序列的各阶元预测信息。我们以准确性(采用sensitivity和F1-score两个指标; Gardner et al, 2019; Hleap et al, 2021)为主, 兼顾运行速度和内存资源占用, 对各个软件的性能进行了综合比较和评估。我们同时选取了4类代表性土壤动物: 弹尾纲(Collembola, 即跳虫)、蜱螨亚纲(Acari, 即蜱虫和螨虫)、环带纲(Clitellata, 即蚯蚓和蛭等)和色矛纲(Chromadorea, 即线虫)以表征不同类别、生活习惯和体型的土壤动物, 并分别构建了这些类群的COI、16S和18S数据库。软件准确性(科、属阶元水平)的比较和评估基于这些参考数据库展开。
1 材料与方法
1.1 数据库构建
本研究选取了弹尾纲、蜱螨亚纲、环带纲和色矛纲等4个类群作为土壤动物代表类群。这4个类群包含了节肢动物、环节动物和线虫动物, 囊括了不同的体型大小和生活习性, 且在土壤动物研究中比较热门, 具有代表性。
我们为4类土壤动物代表类群构建了各自的参考数据库, 包括COI (标准的658 bp片段)、16S (V4‒V5片段, 约400 bp)和18S (V4片段, 约400 bp)的数据库。为了评估各个分子分类预测软件在更复杂的群落中的表现, 我们把4个类群的COI数据库组合并构建了一个额外的COI多类群数据库。由于分子分类预测的运行时间和计算资源损耗与数据库的大小显著相关, 因此在数据库中每个物种的序列只保留1条, 除非存在同种相似度差异较大(10%以上)的序列。同时, 我们会将每一条序列与同一类群中的其他序列进行比对, 相似度普遍较低的序列会被标记为可疑序列并在系统发育树上进一步观察。所有的序列均来自GenBank (NCBI)数据库和BOLDSystems (
数据库中序列的分类信息从NCBI和BOLDSystems数据库中提取, 并经过目、科、属层级的系统发育树和相似度验证。尽管无法保证分类信息的完全正确, 但本研究通过多种办法尽量确保分类信息的准确性。分类信息的可能错误主要来源于3种情况: (1) NCBI和BOLDSystems间同一类群分类信息的不一致。这种情况较少, 本研究统一根据文献采用最新的分类信息。例如Lumbricidae, 我们依据Chelkha等(2020)将其归于Haplotaxida。(2)两条或多条序列一致(或高度相似)但分类信息不一致。这种情况往往是物种分类信息存在变动或者其中一条或多条序列分类信息有误导致。如果是物种分类信息存在变动, 则采用最新的分类信息。如果物种分类信息不存在变动(即其中一条或多条序列可能存在错误), 则根据系统发育树和相似度综合判断, 如依旧难以确认, 则根据序列在源数据库中的丰度来判断。序列分类信息不一致主要集中在物种水平, 而本研究的目标阶元是科和属, 如果仅是物种水平的分类信息错误极少影响本研究的预测准确性评估。(3)单条序列的分类信息错误。这种错误最难避免, 本研究主要通过两种手段来减少这种错误。其一, 将每条序列与其所属的属、科、目阶元的其他序列各进行一次系统发育树和相似度验证。其二, 每个物种只保留丰度最高的一条序列, 即这条序列是该物种中最可靠的序列。
1.2 分子分类预测软件
我们选取了5款主流的分子分类预测软件。这些软件具有较高的引用率, 拥有详细的使用教程和手册, 而且对用户免费开放。其中两款基于相似度算法: HS-BLASTN v0.0.5 + (Chen et al, 2015) (在算法逻辑与BLAST相同的基础上优化了运行速度)和VSEARCH v2.13.6 (Rognes et al, 2016); 另外3款基于系统发育位置PP算法: EPA-NG v0.3.7 (Barbera et al, 2019), RAPPAS v1.21 (Linard et al, 2019)和APPLES v2.0.0 (Balaban et al, 2020)。其中, 基于系统发育位置算法的系统发育树由FastTree2 v2.1.10 (Price et al, 2010)构建, 并在下游分析中借助于GAPPA v0.7.1实现预测结果的可视化(Czech et al, 2020)。
1.3 准确性的比较和评估
本研究所涉及的准确性的比较和评估基于现有的分类系统与系统发育研究而获得, 小部分物种中存在争议的科、属等阶元的确立综合了权威性杂志的分类划分、系统发育树判定和相似度判定。
我们把5款分子分类预测软件的运行代码嵌入了自制脚本“classify.sh”中, 并为该脚本设置了一些参数供使用者选择, 其中, 序列对齐参数借助于软件PAPARA v2.5 (Berger & Stamatakis, 2011)。脚本会在最终根据所选择的分类预测软件输出所有目标序列的各阶元预测信息以及可信值(如相似度)。我们基于1.1中得到的共计13个数据库, 对软件的准确性进行比较和评估。对于每个数据库, 我们进行100次重复, 每次从数据库中随机抽取100条序列(如果数据库序列数较少就适当减少)作为目标序列, 剩下的序列作为临时参考数据库, 然后用各个分类预测软件凭借“临时参考数据库”去预测“目标序列”的阶元信息(本研究以科和属为主), 并得到各个软件的TP (真阳性, 指同时存在于目标序列和预测结果中), FP (假阳性, 指在目标序列中并不存在, 却在预测结果中存在), FN (假阴性, 指在目标序列中存在, 却在预测结果中消失)。根据TP、FP和FN可计算得出TPR (true positive rate, 真阳率)和PPV (positive predictive value, 阳性预测值):
其中, sensitivity指标能体现分类预测软件在混合样品或模拟社群中实现准确预测的概率, 而F1-score指标弥补了其不能凸显结果中错误预测分布情况的缺陷。
在软件的准确性比较和评估阶段, 我们用到了“doPick.sh”和“doCheck.sh”两个脚本。前一个脚本用于目标序列的抽取, 临时参考数据库的构建以及其他参考文件的同步; 后一个脚本调用了“classify.sh”, 用来实现所有预测软件的分类预测并同时得到它们的TP、FP和FN。软件准确性比较和评估所用到的脚本可在
1.4 运行速度和内存占用的比较
软件运行速度和内存占用的比较基于两种不同规模和复杂度的参考数据库展开。一个为序列数大于1,000的弹尾纲COI数据库, 另一个为序列数大于5,000的COI多类群数据库。我们用NCBI上下载的7,326条序列作为目标序列进行测试。在运行速度的比较中, 我们把运行时间折算成相对运行速度(以HS-BLASTN在线程数为1时的速度为单位速度), 分别计算出各个软件在线程数为1、4、8、16和32时的相对运行速度。在内存占用的比较中, 我们记录每个软件(单线程)进行预测时的RSS (resident set size)。所有软件测试都在乌班图18.04.1服务器(128 Gb内存, 16核/32线程)中运行。
2 结果
2.1 数据库构建
我们为4类土壤动物代表类群分别构建了COI、16S和18S的参考数据库(经过重复和错误序列的过滤, 最终每个种只保留一条最可靠的序列)。其中, COI数据库的生物多样性最为丰富, 4个类群(按弹尾纲、蜱螨亚纲、环带纲、色矛纲的顺序排列, 下同)分别筛选得到1,211、1,675、1,297和939条序列(即物种数), 并将它们混合得到COI多类群数据库(5,122条序列)。16S数据库的物种、属和科阶元的丰富度都比较低, 除了环带纲(物种、属和科阶元数分别为972、214和28, 表1)。18S数据库的属和科阶元丰富度比较高, 但物种数(分别为163、635、342和1,042, 表1)没有COI数据库丰富。
表1 各个类群COI、16S和18S参考数据库中的物种、属和科阶元的数目
Table 1
| 类群 Taxa | 分子标记 Markers | 物种数目 Species number | 属数目 Genus number | 科数目 Family number |
|---|---|---|---|---|
| 弹尾纲 Collembola | COI | 1,211 | 157 | 22 |
| 16S | 387 | 81 | 18 | |
| 18S | 163 | 79 | 19 | |
| 蜱螨亚纲 Acari | COI | 1,675 | 460 | 190 |
| 16S | 456 | 76 | 28 | |
| 18S | 635 | 459 | 220 | |
| 环带纲 Clitellata | COI | 1,297 | 255 | 39 |
| 16S | 972 | 214 | 28 | |
| 18S | 342 | 203 | 42 | |
| 色矛纲 Chromadorea | COI | 939 | 249 | 86 |
| 16S | 170 | 64 | 28 | |
| 18S | 1,042 | 430 | 135 | |
| 4个类群合并 Merged | COI | 5,122 | 1,121 | 337 |
我们以COI为例, 将过滤后的精简版参考数据库与未过滤前的原始数据库进行运行时间上的比较。我们以100条COI序列作为目标序列, 使用单个线程的VSEARCH进行分类预测。结果表明, 弹尾纲和蜱螨亚纲的精简版参考数据库节省了80倍以上的时间, 环带纲和色矛纲节省了20倍左右的时间(附录2)。
2.2 准确性的比较和评估
2.2.1 COI数据库
对于COI数据库, 当采用sensitivity指标时, EPA-NG的表现最为出色, 且优势极为明显, 无论是科阶元(0.96 ± 0.02、0.86 ± 0.03、0.93 ± 0.03和0.88 ± 0.03; 按弹尾纲、蜱螨亚纲、环带纲、色矛纲的顺序排列, 下同)还是属阶元(0.74 ± 0.04、0.73 ± 0.04、0.70 ± 0.05和0.71 ± 0.04) (图1a‒d, 附录4)。其次依次是HS-BLASTN (科: 0.84 ± 0.03、0.73 ± 0.05、0.86 ± 0.03和0.77 ± 0.04; 属: 0.64 ± 0.05、0.62 ± 0.05、0.58 ± 0.05和0.60 ± 0.05)和VSEARCH (科: 0.77 ± 0.04、0.71 ± 0.05、0.84 ± 0.04和0.77 ± 0.03; 属: 0.59 ± 0.04、0.60 ± 0.05、0.56 ± 0.06和0.57 ± 0.04), 而RAPPAS和APPLES的sensitivity指标较低(图1a‒d, 附录4)。
图1
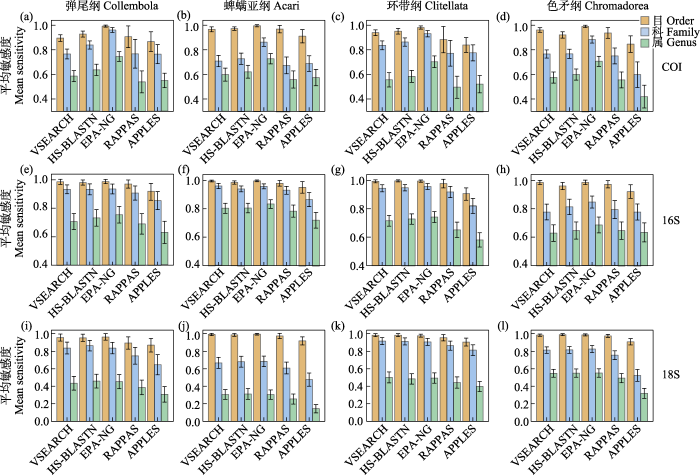
图1
5款分类预测软件在4个类群和3种分子标记应用中的平均敏感度
Fig. 1
Mean sensitivity of five taxonomic assignment tools among four groups and three markers
当采用F1-score指标时, EPA-NG依旧保持着显著优势(科: 0.96 ± 0.02、0.87 ± 0.03、0.93 ± 0.02和0.89 ± 0.03; 属: 0.75 ± 0.04、0.74 ± 0.04、0.72 ± 0.05和0.73 ± 0.04, 图2a‒d, 附录4)。APPLES (科: 0.85 ± 0.05、0.78 ± 0.05、0.86 ± 0.04和0.73 ± 0.09; 属: 0.67 ± 0.05、0.68 ± 0.06、0.64 ± 0.06和0.56 ± 0.09, 图2a‒d, 附录4)在该指标下的表现超越了HS-BLASTN和VSEARCH, 尽管在预测色矛纲时的表现依旧不够亮眼。
图2
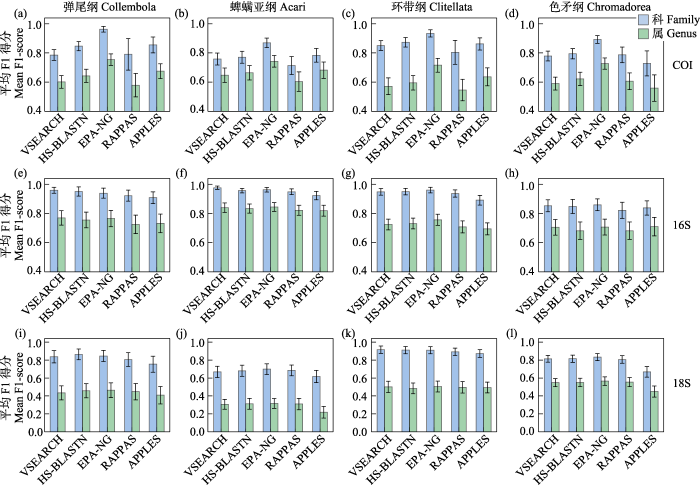
图2
5款分类预测软件在4个类群和3种条码应用中的平均F1分数
Fig. 2
Mean F1-score of five taxonomic assignment tools among four groups and three markers
2.2.2 16S数据库
对于16S数据库, 当采用sensitivity指标时, 尽管EPA-NG (科: 0.94 ± 0.03、0.96 ± 0.02、0.96 ± 0.02和0.85 ± 0.04; 属: 0.76 ± 0.06、0.83 ± 0.03、0.74 ± 0.04和0.69 ± 0.06, 图1e‒h, 附录4)依旧位居首位, 但优势远不及应用COI数据库时。HS-BLASTN (0.93 ± 0.04、0.94 ± 0.02、0.95 ± 0.02和0.81 ± 0.05; 属: 0.73 ± 0.06、0.80 ± 0.03、0.73 ± 0.04和0.65 ± 0.06)和VSEARCH (0.93 ± 0.03、0.96 ± 0.02、0.94 ± 0.02和0.78 ± 0.06; 属: 0.71 ± 0.06、0.80 ± 0.04、0.72 ± 0.04和0.63 ± 0.06)紧随其后(图1e‒h, 附录4)。另外, RAPPAS在应用16S时sensitivity指标比APPLES表现更佳(图1e‒h)。
当采用F1-score指标时, 在科阶元上, VSEARCH (0.96 ± 0.02、0.98 ± 0.01、0.95 ± 0.02和0.85 ± 0.04), EPA-NG (0.94 ± 0.03、0.96 ± 0.02、0.96 ± 0.02和0.86 ± 0.04)和HS-BLASTN (0.95 ± 0.03、0.96 ± 0.02、0.95 ± 0.02和0.85 ± 0.05)的表现都很出色, 且十分接近; 在属阶元上, EPA-NG (0.77 ± 0.06、0.84 ± 0.03、0.76 ± 0.04和0.71 ± 0.05)以轻微的优势位居第一, 其次是VSEARCH (0.77 ± 0.05、0.84 ± 0.03、0.72 ± 0.04和0.70 ± 0.05)和HS-BLASTN (0.75 ± 0.05、0.83 ± 0.03、0.73 ± 0.04和0.68 ± 0.06) (图2e‒h, 附录4)。
2.2.3 18S数据库
对于18S数据库, 当采用sensitivity指标时, EPA-NG (科: 0.84 ± 0.06、0.69 ± 0.06、0.91 ± 0.04和0.83 ± 0.04; 属: 0.45 ± 0.08、0.30 ± 0.05、0.49 ± 0.06和0.55 ± 0.05), HS-BLASTN (科: 0.86 ± 0.06、0.68 ± 0.06、0.91 ± 0.04和0.81 ± 0.04; 属: 0.46 ± 0.08、0.31 ± 0.06、0.48 ± 0.06和0.55 ± 0.05)和VSEARCH (科: 0.84 ± 0.07、0.67 ± 0.06、0.92 ± 0.04和0.81 ± 0.04; 属: 0.43 ± 0.08、0.31 ± 0.06、0.50 ± 0.06和0.55 ± 0.05)的表现十分接近, EPA-NG不再存在明显优势(图1i‒l, 附录4)。同16S, RAPPAS在应用18S时在sensitivity指标上优于APPLES (图1i‒l)。
当采用F1-score指标时, APPLES相对落后, 其他4款软件F1-score值都十分接近(图2i‒l, 附录4)。
2.2.4 COI多类群数据库
对于4个类群的COI序列组成的额外数据库, 趋势和单个类群的COI数据库基本相同, 但APPLES表现得更为乏力。当采用sensitivity指标时, 按排名顺序依次是EPA-NG (科: 0.90 ± 0.02; 属: 0.71 ± 0.03)、HS-BLASTN (科: 0.79 ± 0.03; 属: 0.62 ± 0.04)、VSEARCH (科: 0.76 ± 0.03; 属: 0.58 ± 0.03)、RAPPAS (科: 0.73 ± 0.05; 属: 0.53 ± 0.05)和APPLES (科: 0.65 ± 0.07; 属: 0.49 ± 0.06) (图3a, 附录4)。当采用F1-score指标时, 按排名顺序依次是EPA-NG (科: 0.91 ± 0.02; 属: 0.73 ± 0.03)、HS-BLASTN (科: 0.82 ± 0.03; 属: 0.64 ± 0.03)、VSEARCH (科: 0.79 ± 0.03; 属: 0.61 ± 0.03)、APPLES (科: 0.76 ± 0.05; 属: 0.62 ± 0.05)和RAPPAS (科: 0.77 ± 0.04; 属: 0.58 ± 0.05) (图3b, 附录4)。
图3
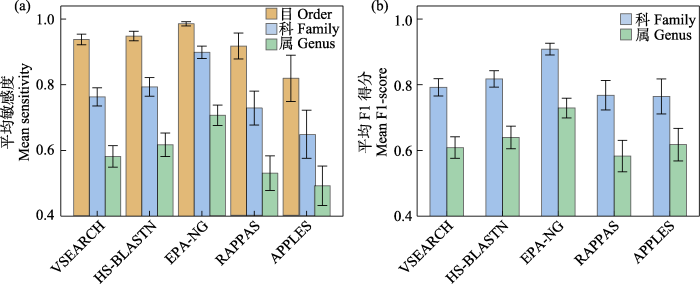
图3
5款分类预测软件在混合COI参考数据库应用中的平均敏感度(a)和平均F1分数(b)
Fig. 3
Mean sensitivity (a) and mean F1-score (b) of five taxonomic assignment tools with merged COI reference database
2.3 内存占用和运行速度的比较
不同分子分类预测软件的计算资源(内存)占用和运行时间损耗存在较大差异。
我们把HS-BLASTN在单个线程时完成7,326条目标序列时的速度设为单位速度, 该过程损耗时间为91.6 s (137 s; 前后分别代表在应用大于1,000和大于5,000序列数的数据库时的时间损耗, 下同) (图4a, c, 附录3)。RAPPAS是唯一一款速度不随着线程数的增加而增加的软件, 无论使用多少线程, 它的运行时间都是172 s (400 s) (图4a, c, 附录3)。此外, RAPPAS也是唯一一款运行速度会随着鉴定次数缓慢加快的软件(附录1), 这可能运用了缓存或锻炼技术。VSEARCH是运行速度最快的软件, 它在单线程和24线程时运行时间分别为8.5 s和1 s (13.9 s和1.3 s) (图4a, c, 附录3)。其次是EPA-NG, 在单线程和24线程时运行时间分别为24 s和2.8 s (91.6 s和11.2 s) (图4a, c, 附录3)。然而, 在应用大于5,000序列数的数据库时, 线程数超过16的情况下, HS-BLASTN运行速度比EPA-NG更快(图4a, c)。APPLES是除RAPPAS之外运行速度最慢的, 在单线程和24线程时运行时间分别为719 s和50.9 s (3,000 s和208 s) (图4a, c, 附录3)。另外, 软件运行速度随线程数增加而加快的现象在线程数大于16时逐渐趋于不明显。
图4
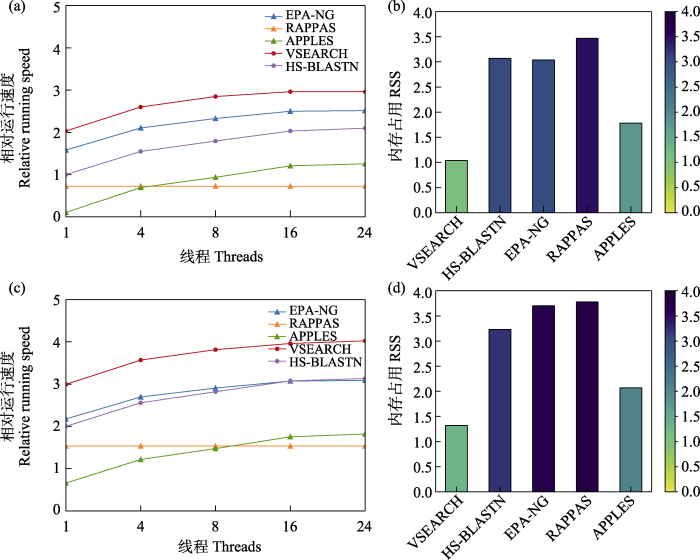
图4
5款分类预测软件在1,000量级(a, b)和5,000量级(c, d)参考数据库应用中的(相对)运行速度和内存占用
Fig. 4
Relative running speed and memory usage of five taxonomic assignment tools when applying reference databases with 1,000 sequences (a and b) and 5,000 sequences (c and d) respectively
3 讨论
3.1 土壤动物的分子分类预测
在本研究中, 我们模拟了在参考数据库缺少目标物种的场景中对土壤动物进行分子鉴定, 结果发现即使是在这种前提下, 5款分子分类预测软件都能将大部分土壤动物鉴定至科级阶元(附录4)。但由于参考数据库缺乏部分目标物种对应的属阶元, 且部分物种属阶元的确立因分类和鉴定工作的不完善(傅声雷, 2007)等原因导致至今仍存在争议, 成功恢复到属阶元的比例要一定程度低于恢复到科级阶元的比例。除本研究外, 郝金凤等(2017)、Hardulak等(2020)和Wang等(2018)分别将分子分类预测应用于蝗虫、甲虫和蚂蚁等并有效实现了这些类群的分子鉴定。总得来说, 分子分类预测能够准确高效地应用于参考数据库匮乏的土壤动物领域, 如果目标是参考数据库中缺乏的物种, 可以凭借近缘物种预测到属或者科阶元。
3.2 分子标记的选择
土壤动物的生物多样性极其丰富, 但其条形码数据库高度不完整, 缺乏大量物种(Decaëns, 2010)。总体来说, 对于物种水平, COI参考数据库丰富度最高; 而对于属和科水平, COI和18S参考数据库的丰富度都较16S更高。对于同一条形码, 各个类群的丰富度也存在差异。例如, 弹尾纲的18S数据库丰富度相对其他类群较低; 环带纲的16S数据库丰富度相对其他类群较高。
分子标记的选择很大程度上会影响基于宏条形码技术的调查和监测结果(Clarke et al, 2017)。在动物界, 凭借着拥有更多高质量的参考序列, 658 bp的COI片段被公认为标准DNA条形码(Hebert et al, 2003; Rodgers et al, 2017)。然而, Deagle等(2014)则认为COI片段过低的保守性会导致一些亲缘关系较近的物种间存在较低的相似性, 因此一些研究尝试使用更保守的16S或18S片段作为分子标记进行探究(Yang, 2013; Elbrecht et al, 2016)。在我们的研究中, 在应用16S时, 各个软件准确性层面的两个指标值都比较高, 这在一定程度上能反映16S在分类预测上的优势。然而, 在种、属和科3个水平上, 16S条形码的丰富度都十分匮乏(Braukmann et al, 2019)。
3.3 准确性的比较和评估
不同软件间的准确性存在差异, 相同软件在不同宏条形码片段或不同类群的数据库中也有不同的表现(Brandt et al, 2021)。在使用同一宏条形码片段时, 尽管5款所选的软件各自在不同类群中的表现存在差异, 但它们的排名趋势在不同类群中基本一致。各软件间的差异在不同条形码类型中有不同程度的体现。各软件间的准确性差异在COI条形码中最显著。在应用COI条形码时, EPA-NG在属和科水平上的准确性比其他软件有显著优势, 这种显著优势同时表现在sensitivity和F1-score两个指标上, 表明EPA-NG在应用COI条形码时同时具备较低的假阴性和假阳性。APPLES在sensitivity指标上不如HS-BLASTN和VSEARCH, 但在F1-score上表现比HS-BLASTN和VSEARCH更出色(除了色矛纲)。这体现了APPLES在应用COI条形码时拥有较低的假阳性(Balaban et al, 2020)和较高的假阴性, 这可能与它保守的算法有关, 不容易出现鉴定错误也不容易将序列鉴定到低阶元。另外, 在参考数据库加大的情况下, APPLES的准确性表现严重不稳定。在应用16S条形码时, 软件间的准确性差异远不及应用COI时。在使用sensitivity指标时, EPA-NG在属和科水平上的准确性表现都最出色, 尽管与其他软件的差距较应用COI时大大减小; 在使用F1-score指标时, EPA-NG、VSEARCH和HS-BLASTN都表现优异, 且差距极小。在应用18S条形码时, 各软件的准确性差异进一步缩小。EPA-NG、VSEARCH和HS- BLASTN的表现十分接近, 在使用F1-score指标时, RAPPAS的准确性也趋近于它们。
总的来说, EPA-NG的准确性表现在不同分子标记间最稳定, 始终保持着较高的准确性, 尤其是在使用COI条形码时, 表现出显著优势。同其他文章中软件比较和评估的结果一样(Brandt et al, 2021; Hleap et al, 2021), BLAST (本研究中使用HS- BLASTN代替, HS-BLASTN是快速版本的BLAST; Chen et al, 2015)在不同分子标记和类群中始终有较为稳定的出色表现(Bik et al, 2021; Hleap et al, 2021)。作为USEARCH的高质量开源替代, VSEARCH的准确性与BLAST十分接近, 尽管假阳性略高于BLAST (Edgar, 2010; Rognes et al, 2016)。APPLES的算法可能比较保守, 更适用于较高的分类水平(目及目以上) RAPPAS在准确性上表现并不突出, 其在免对齐上的优势在本研究中无法体现是非常可惜的。
3.4 其他性能的比较和评估
在运行速度上, 各个软件都能在数秒到数分钟内完成成千上万条序列的分类预测。尤其是VSEARCH、EPA-NG和HS-BLASTN, 在线程足够的情况下, 可以瞬间完成大批量鉴定。其中, VSEARCH的运行速度最快, 反映出其在算法上的优势(Rognes et al, 2016)。除RAPPAS之外, 其他软件的运行速度都随着线程的增加而加快, 但当线程数大于16时, 运行速度的增加并不显著。RAPPAS速度的优化可能通过缓存或者锻炼来实现, 它的运行速度会随着分类预测次数的增加而缓慢加快。随着参考数据库序列数增加, 分类预测的运行速度会随之减慢(Hleap et al, 2021), 此时基于相似度算法的软件比基于系统发育位置算法的软件运行速度损耗更小, 因此当参考数据库过大时, 基于系统发育位置算法的软件可能更加费时。
在内存占用上, VSEARCH的内存占用最小, 其次是APPLES, 它们的内存占用远小于其他3款软件, 尽管它们的内存占用会随着线程的增加而增加。EPA-NG随着参考数据库大小的增加内存占用也大大增加, 这可能会限制其在大型数据库应用中的发挥。除EPA-NG外, 其他软件在参考数据库大小增长至原来5倍左右的情况下, 内存占用增长在一倍以内, 这得益于算法在计算资源损耗上的不断优化。
总的来说, VSEARCH的运行速度最快, 内存占用最小。EPA-NG在参考数据库较小时运行时间和计算资源的损耗较少, 但当参考数据库较大时, 这些损耗会限制EPA-NG的发挥。HS-BLASTN在参考数据库较小时比EPA-NG损耗更多的运行时间和计算资源, 但当参考数据库较大时, 它比EPA-NG更节省运行时间和计算资源。APPLES内存占用较小, 但运行速度较慢。RAPPAS作为一款不需要对齐的基于系统发育位置算法的软件(Linard et al, 2019), 在不需要对齐的同时, 也付出了需要更多运行时间和内存占用的代价。
3.5 总结和展望
和大部分动物类群一样, 在4个土壤动物类群中, COI参考数据库的物种种类最繁多, 属和科水平的丰富度也很高, 这是很多动物领域的宏条形码研究选用COI作为分子标记的主要原因之一(Braukmann et al, 2019)。在应用COI条形码时, EPA- NG的准确性显著高于其他4款软件, 但随着参考数据库大小的增加, 运行时间损耗和内存占用增幅也比其他软件显著。因此, 在参考数据库不庞大的前提下(例如, 针对单个类群或类群较少时), EPA-NG是应用COI条形码时的最佳首选; 但当数据库足够庞大时(例如, 针对整个动物界或类群较多时), 运行时间和内存占用更节省的基于相似度算法的软件更具竞争优势(表2)。VSEARCH和HS-BLASTN准确性十分接近, 但是无论是在运行时间还是在内存占用上, VSEARCH的表现都更加出色(Rognes et al, 2016)。因此, 越来越多的应用宏条形码技术的研究报告使用VSEARCH来代替BLAST进行分子鉴定(Cavaliere et al, 2021; Lanzén et al, 2021; Torrell et al, 2021)。此外, VSEARCH还自带一些过滤、去噪和归一化等功能(Rognes et al, 2016), 所以本研究也同样推荐VSEARCH来取代BLAST。
表2 5款分类预测软件的推荐使用情况
Table 2
| 分类预测软件 Tools | 推荐使用情况 Recommendation on application |
|---|---|
| EPA-NG | 以COI作为分子标记且参考数据库不大的场合 COI is used as the marker and the reference database is small |
| VSEARCH | 以16S或18S作为分子标记或者参考数据库较大的场合 16S/18S is used as the marker; the reference database includes thousands of sequences or more |
| HS-BLASTN | 同VSEARCH, 但优先级不如VSEARCH Similar to VSEARCH |
| APPLES | 仅预测较高阶元的场合 Predicting higher taxonomical hierarchy |
| RAPPAS | 目标序列间长度差异较大的场合 When sequence lengths differ greatly |
单个条形码都有着它自身的局限性, 因此条形码间的组合使用以实现各自的优势互补或许会成为将来宏条形码技术的主流趋势(张宛宛等, 2017), 例如COI + 16S或COI + 18S等。现如今, 分子分类预测方法层出不尽, 除了主流的相似度算法和系统发育树放置算法, 其他算法也逐渐被开发出来, 例如基于机器学习的算法(Murali et al, 2018)等。将来的土壤动物分类预测可以不断去尝试这些新鲜的算法。另外, 相似度算法和系统发育位置算法以及其他算法都有自身的优劣, 将来或许可以尝试将两个或多个算法结合起来, 实现更精准更高效的分子分类预测, 例如EPA-NG和VSEARCH的组合等。
本文比较和评估了目前几款主流的分子分类预测软件在土壤动物应用中的性能, 虽然不能囊括所有的分类预测软件, 但希望能为今后基于宏条形码等技术的土壤动物调查和监测提供一些借鉴意义。
Supplementary Material
附录1 RAPPAS运行时间随数据库使用次数变化情况
Appendix 1 Variation of running time for RAPPAS during usage count of the reference database
附录2 过滤前后参考数据库分类预测耗时比较
Appendix 2 Running time comparison between the databases before and after filtering
附录3 5款分类预测软件在1,000量级和5,000量级参考数据库应用中的运行时间和内存占用
Appendix 3 Running time and memory usage of five taxonomic assignment tools when applying reference databases with 1,000 sequences and 5,000 sequences respectively
附录4 5款分类预测软件在各个类群和条码应用中的准确性具体表现
Appendix 4 Accuracy performance of five taxonomic assignment tools among different groups and different markers
参考文献
Basic local alignment search tool
DOI:10.1016/S0022-2836(05)80360-2
PMID:2231712
[本文引用: 1]

A new approach to rapid sequence comparison, basic local alignment search tool (BLAST), directly approximates alignments that optimize a measure of local similarity, the maximal segment pair (MSP) score. Recent mathematical results on the stochastic properties of MSP scores allow an analysis of the performance of this method as well as the statistical significance of alignments it generates. The basic algorithm is simple and robust; it can be implemented in a number of ways and applied in a variety of contexts including straightforward DNA and protein sequence database searches, motif searches, gene identification searches, and in the analysis of multiple regions of similarity in long DNA sequences. In addition to its flexibility and tractability to mathematical analysis, BLAST is an order of magnitude faster than existing sequence comparison tools of comparable sensitivity.
Metabarcoding and mitochondrial metagenomics of endogean arthropods to unveil the mesofauna of the soil
DOI:10.1111/2041-210X.12557 URL [本文引用: 1]
The limited spatial scale of dispersal in soil arthropods revealed with whole-community haplotype- level metabarcoding
DOI:10.1111/mec.15591 URL [本文引用: 2]
APPLES: Scalable distance-based phylogenetic placement with or without alignments
DOI:10.1093/sysbio/syz063
PMID:31545363
[本文引用: 2]
Placing a new species on an existing phylogeny has increasing relevance to several applications. Placement can be used to update phylogenies in a scalable fashion and can help identify unknown query samples using (meta-)barcoding, skimming, or metagenomic data. Maximum likelihood (ML) methods of phylogenetic placement exist, but these methods are not scalable to reference trees with many thousands of leaves, limiting their ability to enjoy benefits of dense taxon sampling in modern reference libraries. They also rely on assembled sequences for the reference set and aligned sequences for the query. Thus, ML methods cannot analyze data sets where the reference consists of unassembled reads, a scenario relevant to emerging applications of genome skimming for sample identification. We introduce APPLES, a distance-based method for phylogenetic placement. Compared to ML, APPLES is an order of magnitude faster and more memory efficient, and unlike ML, it is able to place on large backbone trees (tested for up to 200,000 leaves). We show that using dense references improves accuracy substantially so that APPLES on dense trees is more accurate than ML on sparser trees, where it can run. Finally, APPLES can accurately identify samples without assembled reference or aligned queries using kmer-based distances, a scenario that ML cannot handle. APPLES is available publically at github.com/balabanmetin/apples.© The Author(s) 2019. Published by Oxford University Press, on behalf of the Society of Systematic Biologists. All rights reserved. For permissions, please email: journals.permissions@oup.com.
Accuracy, limitations and cost efficiency of eDNA-based community survey in tropical frogs
DOI:10.1111/1755-0998.12934
PMID:30155977
[本文引用: 1]
Rapid environmental change in highly biodiverse tropical regions demands efficient biomonitoring programmes. While existing metrics of species diversity and community composition rely on encounter-based survey data, eDNA recently emerged as alternative approach. Costs and ecological value of eDNA-based methods have rarely been evaluated in tropical regions, where high species richness is accompanied by high functional diversity (e.g., the use of different microhabitats by different species and life stages). We first tested whether estimation of tropical frogs' community structure derived from eDNA data is compatible with expert field assessments. Next, we evaluated whether eDNA is a financially viable solution for biodiversity monitoring in tropical regions. We applied eDNA metabarcoding to investigate frog species occurrence in five ponds in the Chiquitano dry forest region in Bolivia and compared our data with a simultaneous visual and audio encounter survey (VAES). We found that taxon lists and community structure generated with eDNA and VAES correspond closely, and most deviations are attributable to different species' life histories. Cost efficiency of eDNA surveys was mostly influenced by the richness of local fauna and the number of surveyed sites: VAES may be less costly in low-diversity regions, but eDNA quickly becomes more cost-efficient in high-diversity regions with many sites sampled. The results highlight that eDNA is suitable for large-scale biodiversity surveys in high-diversity areas if life history is considered, and certain precautions in sampling, genetic analyses and data interpretation are taken. We anticipate that spatially extensive, standardized eDNA biodiversity surveys will quickly emerge in the future.© 2018 John Wiley & Sons Ltd.
EPA-ng: Massively parallel evolutionary placement of genetic sequences
DOI:10.1093/sysbio/syy054
PMID:30165689
[本文引用: 1]
Next generation sequencing (NGS) technologies have led to a ubiquity of molecular sequence data. This data avalanche is particularly challenging in metagenetics, which focuses on taxonomic identification of sequences obtained from diverse microbial environments. Phylogenetic placement methods determine how these sequences fit into an evolutionary context. Previous implementations of phylogenetic placement algorithms, such as the evolutionary placement algorithm (EPA) included in RAxML, or PPLACER, are being increasingly used for this purpose. However, due to the steady progress in NGS technologies, the current implementations face substantial scalability limitations. Herein, we present EPA-NG, a complete reimplementation of the EPA that is substantially faster, offers a distributed memory parallelization, and integrates concepts from both, RAxML-EPA and PPLACER. EPA-NG can be executed on standard shared memory, as well as on distributed memory systems (e.g., computing clusters). To demonstrate the scalability of EPA-NG, we placed $1$ billion metagenetic reads from the Tara Oceans Project onto a reference tree with 3748 taxa in just under $7$ h, using 2048 cores. Our performance assessment shows that EPA-NG outperforms RAxML-EPA and PPLACER by up to a factor of $30$ in sequential execution mode, while attaining comparable parallel efficiency on shared memory systems. We further show that the distributed memory parallelization of EPA-NG scales well up to 2048 cores. EPA-NG is available under the AGPLv3 license: https://github.com/Pbdas/epa-ng.
Belowground biodiversity and ecosystem functioning
DOI:10.1038/nature13855 URL [本文引用: 1]
A comparative evaluation of sequence classification programs
DOI:10.1186/1471-2105-13-92
PMID:22574964
[本文引用: 1]
Background: A fundamental problem in modern genomics is to taxonomically or functionally classify DNA sequence fragments derived from environmental sampling (i.e., metagenomics). Several different methods have been proposed for doing this effectively and efficiently, and many have been implemented in software. In addition to varying their basic algorithmic approach to classification, some methods screen sequence reads for 'barcoding genes' like 16S rRNA, or various types of protein-coding genes. Due to the sheer number and complexity of methods, it can be difficult for a researcher to choose one that is well-suited for a particular analysis.;Results: We divided the very large number of programs that have been released in recent years for solving the sequence classification problem into three main categories based on the general algorithm they use to compare a query sequence against a database of sequences. We also evaluated the performance of the leading programs in each category on data sets whose taxonomic and functional composition is known.;Conclusions: We found significant variability in classification accuracy, precision, and resource consumption of sequence classification programs when used to analyze various metagenomics data sets. However, we observe some general trends and patterns that will be useful to researchers who use sequence classification programs.
Performance, accuracy, and web server for evolutionary placement of short sequence reads under maximum likelihood
DOI:10.1093/sysbio/syr010
PMID:21436105
[本文引用: 1]
We present an evolutionary placement algorithm (EPA) and a Web server for the rapid assignment of sequence fragments (short reads) to edges of a given phylogenetic tree under the maximum-likelihood model. The accuracy of the algorithm is evaluated on several real-world data sets and compared with placement by pair-wise sequence comparison, using edit distances and BLAST. We introduce a slow and accurate as well as a fast and less accurate placement algorithm. For the slow algorithm, we develop additional heuristic techniques that yield almost the same run times as the fast version with only a small loss of accuracy. When those additional heuristics are employed, the run time of the more accurate algorithm is comparable with that of a simple BLAST search for data sets with a high number of short query sequences. Moreover, the accuracy of the EPA is significantly higher, in particular when the sample of taxa in the reference topology is sparse or inadequate. Our algorithm, which has been integrated into RAxML, therefore provides an equally fast but more accurate alternative to BLAST for tree-based inference of the evolutionary origin and composition of short sequence reads. We are also actively developing a Web server that offers a freely available service for computing read placements on trees using the EPA.
Aligning short reads to reference alignments and trees
DOI:10.1093/bioinformatics/btr320
PMID:21636595
[本文引用: 2]
Likelihood-based methods for placing short read sequences from metagenomic samples into reference phylogenies have been recently introduced. At present, it is unclear how to align those reads with respect to the reference alignment that was deployed to infer the reference phylogeny. Moreover, the adaptability of such alignment methods with respect to the underlying reference alignment strategies/philosophies has not been explored. It has also not been assessed if the reference phylogeny can be deployed in conjunction with the reference alignment to improve alignment accuracy in this context.We assess different strategies for short read alignment and propose a novel phylogeny-aware alignment procedure. Our alignment method can improve the accuracy of subsequent phylogenetic placement of the reads into a reference phylogeny by up to 5.8 times compared with phylogeny-agnostic methods. It can be deployed to align reads to alignments generated by using fundamentally different alignment strategies (e.g. PRANK(+F) versus MUSCLE).http://www.exelixis-lab.org/software.html
Just keep it simple? Benchmarking the accuracy of taxonomy assignment software in metabarcoding studies
DOI:10.1111/1755-0998.13473
PMID:34268901
[本文引用: 1]
How do you put a name on an unknown piece of DNA? From microbes to mammals, high-throughput metabarcoding studies provide a more objective view of natural communities, overcoming many of the inherent limitations of traditional field surveys and microscopy-based observations (Deiner et al. 2017). Taxonomy assignment is one of the most critical aspects of any metabarcoding study, yet this important bioinformatics task is routinely overlooked. Biodiversity surveys and conservation efforts often depend on formal species inventories: the presence (or absence) of species, and the number of individuals reported across space and time. However, computational workflows applied in eukaryotic metabarcoding studies were originally developed for use with bacterial/archaeal datasets, where microbial researchers rely on one conserved locus (nuclear 16S rRNA) and have access to vast databases with good coverage across most prokaryotic lineages - a situation not mirrored in most multicellular taxa. In this issue of Molecular Ecology Resources, Hleap et al. (2021) carry out an extensive benchmarking exercise focused on taxonomy assignment strategies for eukaryotic metabarcoding studies utilizing the mitochondrial Cytochrome C Oxidase I marker gene (COI). They assess the performance and accuracy of software tools representing diverse methodological approaches: from "simple" strategies based on sequence similarity and composition, to model-based phylogenetic and probabilistic classification tools. Contrary to popular assumptions, less complex approaches (BLAST and the QIIME2 feature classifier) consistently outperformed more sophisticated mathematical algorithms and were highly accurate for assigning taxonomy at higher levels (e.g. family). Lower-level assignments at the genus and species level still pose significant challenge for most existing algorithms, and sparse eukaryotic reference databases further limit software performance. This study illuminates current best practices for metabarcoding taxonomy assignments, and underscores the need for community-driven efforts to expand taxonomic and geographic representation in reference DNA barcode databases.This article is protected by copyright. All rights reserved.
Performance of amplicon and shotgun sequencing for accurate biomass estimation in invertebrate community samples
DOI:10.1111/1755-0998.12888 URL [本文引用: 1]
Environmental DNA for wildlife biology and biodiversity monitoring
DOI:10.1016/j.tree.2014.04.003
PMID:24821515
[本文引用: 1]
Extraction and identification of DNA from an environmental sample has proven noteworthy recently in detecting and monitoring not only common species, but also those that are endangered, invasive, or elusive. Particular attributes of so-called environmental DNA (eDNA) analysis render it a potent tool for elucidating mechanistic insights in ecological and evolutionary processes. Foremost among these is an improved ability to explore ecosystem-level processes, the generation of quantitative indices for analyses of species, community diversity, and dynamics, and novel opportunities through the use of time-serial samples and unprecedented sensitivity for detecting rare or difficult-to-sample taxa. Although technical challenges remain, here we examine the current frontiers of eDNA, outline key aspects requiring improvement, and suggest future developments and innovations for research. Copyright © 2014 Elsevier Ltd. All rights reserved.
Bioinformatic pipelines combining denoising and clustering tools allow for more comprehensive prokaryotic and eukaryotic metabarcoding
DOI:10.1111/1755-0998.13398
PMID:33835712
[本文引用: 3]
Environmental DNA metabarcoding is a powerful tool for studying biodiversity. However, bioinformatic approaches need to adjust to the diversity of taxonomic compartments targeted as well as to each barcode gene specificities. We built and tested a pipeline based on read correction with DADA2 allowing analysing metabarcoding data from prokaryotic (16S) and eukaryotic (18S, COI) life compartments. We implemented the option to cluster Amplicon Sequence Variants (ASVs) into Operational Taxonomic Units (OTUs) with swarm, a network-based clustering algorithm, and the option to curate ASVs/OTUs using LULU. Finally, taxonomic assignment was implemented via the Ribosomal Database Project Bayesian classifier (RDP) and BLAST. We validate this pipeline with ribosomal and mitochondrial markers using metazoan mock communities and 42 deep-sea sediment samples. The results show that ASVs and OTUs describe different levels of biotic diversity, the choice of which depends on the research questions. They underline the advantages and complementarity of clustering and LULU-curation for producing metazoan biodiversity inventories at a level approaching the one obtained using morphological criteria. While clustering removes intraspecific variation, LULU effectively removes spurious clusters, originating from errors or intragenomic variability. Swarm clustering affected alpha and beta diversity differently depending on genetic marker. Specifically, d-values > 1 appeared to be less appropriate with 18S for metazoans. Similarly, increasing LULU's minimum ratio level proved essential to avoid losing species in sample-poor datasets. Comparing BLAST and RDP underlined that accurate assignments of deep-sea species can be obtained with RDP, but highlighted the need for a concerted effort to build comprehensive, ecosystem-specific databases.This article is protected by copyright. All rights reserved.
Metabarcoding a diverse arthropod mock community
DOI:10.1111/1755-0998.13008
PMID:30779309
[本文引用: 3]
Although DNA metabarcoding is an attractive approach for monitoring biodiversity, it is often difficult to detect all the species present in a bulk sample. In particular, sequence recovery for a given species depends on its biomass and mitome copy number as well as the primer set employed for PCR. To examine these variables, we constructed a mock community of terrestrial arthropods comprised of 374 species. We used this community to examine how species recovery was impacted when amplicon pools were constructed in four ways. The first two protocols involved the construction of bulk DNA extracts from different body segments (Bulk Abdomen, Bulk Leg). The other protocols involved the production of DNA extracts from single legs which were then merged prior to PCR (Composite Leg) or PCR-amplified separately (Single Leg) and then pooled. The amplicons generated by these four treatments were then sequenced on three platforms (Illumina MiSeq, Ion Torrent PGM and Ion Torrent S5). The choice of sequencing platform did not substantially influence species recovery, although the Miseq delivered the highest sequence quality. As expected, species recovery was most efficient from the Single Leg treatment because amplicon abundance varied little among taxa. Among the three treatments where PCR occurred after pooling, the Bulk Abdomen treatment produced a more uniform read abundance than the Bulk Leg or Composite Leg treatment. Primer choice also influenced species recovery and evenness. Our results reveal how variation in protocols can have substantial impacts on perceived diversity unless sequencing coverage is sufficient to reach an asymptote.© 2019 The Authors. Molecular Ecology Resources Published by John Wiley & Sons Ltd.
Assessing the ecological quality status of the highly polluted Bagnoli area (Tyrrhenian Sea, Italy) using foraminiferal eDNA metabarcoding
DOI:10.1016/j.scitotenv.2021.147871 URL [本文引用: 1]
Cutaneous excreta of the earthworm Eisenia fetida (Haplotaxida: Lumbricidae) might hinder the biological control performance of entomopathogenic nematodes
DOI:10.1016/j.soilbio.2019.107691 URL [本文引用: 1]
High speed BLASTN: An accelerated MegaBLAST search tool
DOI:10.1093/nar/gkv784
PMID:26250111
[本文引用: 2]
Sequence alignment is a long standing problem in bioinformatics. The Basic Local Alignment Search Tool (BLAST) is one of the most popular and fundamental alignment tools. The explosive growth of biological sequences calls for speedup of sequence alignment tools such as BLAST. To this end, we develop high speed BLASTN (HS-BLASTN), a parallel and fast nucleotide database search tool that accelerates MegaBLAST--the default module of NCBI-BLASTN. HS-BLASTN builds a new lookup table using the FMD-index of the database and employs an accurate and effective seeding method to find short stretches of identities (called seeds) between the query and the database. HS-BLASTN produces the same alignment results as MegaBLAST and its computational speed is much faster than MegaBLAST. Specifically, our experiments conducted on a 12-core server show that HS-BLASTN can be 22 times faster than MegaBLAST and exhibits better parallel performance than MegaBLAST. HS-BLASTN is written in C++ and the related source code is available at https://github.com/chenying2016/queries under the GPLv3 license.© The Author(s) 2015. Published by Oxford University Press on behalf of Nucleic Acids Research.
A DNA barcoding system integrating multigene sequence data
DOI:10.1111/2041-210X.12366 URL [本文引用: 1]
A protocol for species delineation of public DNA databases, applied to the Insecta
DOI:10.1093/sysbio/syu038
PMID:24929897
[本文引用: 1]
Public DNA databases are composed of data from many different taxa, although the taxonomic annotation on sequences is not always complete, which impedes the utilization of mined data for species-level applications. There is much ongoing work on species identification and delineation based on the molecular data itself, although applying species clustering to whole databases requires consolidation of results from numerous undefined gene regions, and introduces significant obstacles in data organization and computational load. In the current paper, we demonstrate an approach for species delineation of a sequence database. All DNA sequences for the insects were obtained and processed. After filtration of duplicated data, delineation of the database into species or molecular operational taxonomic units (MOTUs) followed a three-step process in which (i) the genetic loci L are partitioned, (ii) the species S are delineated within each locus, then (iii) species units are matched across loci to form the matrix L × S, a set of global (multilocus) species units. Partitioning the database into a set of homologous gene fragments was achieved by Markov clustering using edge weights calculated from the amount of overlap between pairs of sequences, then delineation of species units and assignment of species names were performed for the set of genes necessary to capture most of the species diversity. The complexity of computing pairwise similarities for species clustering was substantial at the cytochrome oxidase subunit I locus in particular, but made feasible through the development of software that performs pairwise alignments within the taxonomic framework, while accounting for the different ranks at which sequences are labeled with taxonomic information. Over 24 different homologs, the unidentified sequences numbered approximately 194,000, containing 41,525 species IDs (98.7% of all found in the insect database), and were grouped into 59,173 single-locus MOTUs by hierarchical clustering under parameters optimized independently for each locus. Species units from different loci were matched using a multipartite matching algorithm to form multilocus species units with minimal incongruence between loci. After matching, the insect database as represented by these 24 loci was found to be composed of 78,091 species units in total. 38,574 of these units contained only species labeled data, 34,891 contained only unlabeled data, leaving 4,626 units composed both of labeled and unlabeled sequences. In addition to giving estimates of species diversity of sequence repositories, the protocol developed here will facilitate species-level applications of modern-day sequence data sets. In particular, the L × S matrix represents a post-taxonomic framework that can be used for species-level organization of metagenomic data, and incorporation of these methods into phylogenetic pipelines will yield matrices more representative of species diversity.© The Author(s) 2014. Published by Oxford University Press, on behalf of the Society of Systematic Biologists. All rights reserved. For Permissions, please email: journals.permissions@oup.com.
Effect of marker choice and thermal cycling protocol on zooplankton DNA metabarcoding studies
DOI:10.1002/ece3.2667
PMID:28168024
[本文引用: 1]
DNA metabarcoding is a promising approach for rapidly surveying biodiversity and is likely to become an important tool for measuring ecosystem responses to environmental change. Metabarcoding markers need sufficient taxonomic coverage to detect groups of interest, sufficient sequence divergence to resolve species, and will ideally indicate relative abundance of taxa present. We characterized zooplankton assemblages with three different metabarcoding markers (nuclear 18S rDNA, mitochondrial COI, and mitochondrial 16S rDNA) to compare their performance in terms of taxonomic coverage, taxonomic resolution, and correspondence between morphology- and DNA-based identification. COI amplicons sequenced on separate runs showed that operational taxonomic units representing >0.1% of reads per sample were highly reproducible, although slightly more taxa were detected using a lower annealing temperature. Mitochondrial COI and nuclear 18S showed similar taxonomic coverage across zooplankton phyla. However, mitochondrial COI resolved up to threefold more taxa to species compared to 18S. All markers revealed similar patterns of beta-diversity, although different taxa were identified as the greatest contributors to these patterns for 18S. For calanoid copepod families, all markers displayed a positive relationship between biomass and sequence reads, although the relationship was typically strongest for 18S. The use of COI for metabarcoding has been questioned due to lack of conserved primer-binding sites. However, our results show the taxonomic coverage and resolution provided by degenerate COI primers, combined with a comparatively well-developed reference sequence database, make them valuable metabarcoding markers for biodiversity assessment.
Genesis and Gappa: Processing, analyzing and visualizing phylogenetic (placement) data
DOI:10.1093/bioinformatics/btaa070
PMID:32016344
[本文引用: 1]
We present genesis, a library for working with phylogenetic data, and gappa, an accompanying command-line tool for conducting typical analyses on such data. The tools target phylogenetic trees and phylogenetic placements, sequences, taxonomies and other relevant data types, offer high-level simplicity as well as low-level customizability, and are computationally efficient, well-tested and field-proven.Both genesis and gappa are written in modern C++11, and are freely available under GPLv3 at http://github.com/lczech/genesis and http://github.com/lczech/gappa.Supplementary data are available at Bioinformatics online.© The Author(s) 2020. Published by Oxford University Press.
DNA metabarcoding and the cytochrome c oxidase subunit I marker: Not a perfect match
DOI:10.1098/rsbl.2014.0562 URL [本文引用: 2]
Macroecological patterns in soil communities
DOI:10.1111/j.1466-8238.2009.00517.x URL [本文引用: 2]
Search and clustering orders of magnitude faster than BLAST
DOI:10.1093/bioinformatics/btq461
PMID:20709691
[本文引用: 1]
Biological sequence data is accumulating rapidly, motivating the development of improved high-throughput methods for sequence classification.UBLAST and USEARCH are new algorithms enabling sensitive local and global search of large sequence databases at exceptionally high speeds. They are often orders of magnitude faster than BLAST in practical applications, though sensitivity to distant protein relationships is lower. UCLUST is a new clustering method that exploits USEARCH to assign sequences to clusters. UCLUST offers several advantages over the widely used program CD-HIT, including higher speed, lower memory use, improved sensitivity, clustering at lower identities and classification of much larger datasets.Binaries are available at no charge for non-commercial use at http://www.drive5.com/usearch.
Testing the potential of a ribosomal 16S marker for DNA metabarcoding of insects
A review and perspective on soil biodiversity research
DOI:10.1360/biodiv.060293
[本文引用: 1]
In this paper, the soil biodiversity and its functioning in ecosystem were briefly summarized, and the history and development of the discipline of soil biology were also reviewed. Meanwhile, I pointed out some issues on soil biology to be addressed for a few years to come. Due to the importance of soil biodiversity to the maintenance of ecosystem functions but inadequate study on soil biota in China, a series of articles on soil biota were collectively published in this issue of <em>Biodiversity Science</em>. The objectives of this issue were to enable the Chinese scientists to better understand the functions of soil biodiversity and to stimulate the interest of young scholars in the discipline of soil biology. The ultimate goal was to push forward the research and development of soil biology in China and to apply the knowledge and techniques of soil biology in the development of national economy.
土壤生物多样性的研究概况与发展趋势
DOI:10.1360/biodiv.060293
[本文引用: 1]
本文概括性地介绍了土壤生物类群的多样性及其在生态系统中的作用; 同时简要地回顾和比较了国内外在土壤生物学方面的研究动态, 分析了土壤生物学今后的发展趋势。鉴于土壤生物在生态系统中的重要性以及我国在土壤生物学研究方面的不足, 《生物多样性》本期刊登了一系列有关土壤生物的文章, 目的是为了使国内科学家对土壤生物多样性在生态系统中的作用有更好的认识, 并希望能够唤起更多的年轻学者加入到土壤生物学研究的行列, 以推动土壤生物学在我国的迅速发展并将土壤生物学的研究成果应用于国民经济的发展中。
Strengthening the research on soil fauna diversity and their ecological functions using novel technology and field experimental facility
DOI:10.17520/biods.2018292 [本文引用: 1]
利用新方法和野外实验平台加强土壤动物多样性及其生态功能的研究
DOI:10.17520/biods.2018292 [本文引用: 1]
Identifying accurate metagenome and amplicon software via a meta-analysis of sequence to taxonomy benchmarking studies
Intraspecific genetic variation in complex assemblages from mitochondrial metagenomics: Comparison with DNA barcodes
DOI:10.1111/2041-210X.12667 URL [本文引用: 1]
Evaluating next-generation sequencing (NGS) methods for routine monitoring of wild bees: Metabarcoding, mitogenomics or NGS barcoding
DOI:10.1111/1755-0998.13013
PMID:30912868
[本文引用: 1]
Implementing cost-effective monitoring programs for wild bees remains challenging due to the high costs of sampling and specimen identification. To reduce costs, next-generation sequencing (NGS)-based methods have lately been suggested as alternatives to morphology-based identifications. To provide a comprehensive presentation of the advantages and weaknesses of different NGS-based identification methods, we assessed three of the most promising ones, namely metabarcoding, mitogenomics and NGS barcoding. Using a regular monitoring data set (723 specimens identified using morphology), we found that NGS barcoding performed best for both species presence/absence and abundance data, producing only few false positives (3.4%) and no false negatives. In contrast, the proportion of false positives and false negatives was higher using metabarcoding and mitogenomics. Although strong correlations were found between biomass and read numbers, abundance estimates significantly skewed the communities' composition in these two techniques. NGS barcoding recovered the same ecological patterns as morphology. Ecological conclusions based on metabarcoding and mitogenomics were similar to those based on morphology when using presence/absence data, but different when using abundance data. In terms of workload and cost, we show that metabarcoding and NGS barcoding can compete with morphology, but not mitogenomics which was consistently more expensive. Based on these results, we advocate that NGS barcoding is currently the seemliest NGS method for monitoring of wild bees. Furthermore, this method has the advantage of potentially linking DNA sequences with preserved voucher specimens, which enable morphological re-examination and will thus produce verifiable records which can be fed into faunistic databases.© 2019 The Authors. Molecular Ecology Resources Published by John Wiley & Sons Ltd.
Diversity investigation and application of DNA barcoding of Acridoidea from Baiyangdian Wetland
DOI:10.17520/biods.2016331
[本文引用: 1]
Both the species diversity and distribution pattern of the superfamily Acridoidea of the suborder Caelifera have important contributions to understanding the local biodiversity of the Baiyangdian Wetland. This research tries to study the species diversity and distribution pattern of the superfamily Acridoidea within the Baiyangdian Wetland and test the feasibility of DNA barcoding in species identification for this superfamily. Sequences of the cox1 gene were obtained from 97 individuals of 21 species of the superfamily Acridoidea. Phylogenetic, genetic distance and sequence difference threshold analyses using the Neighbor Joining (NJ), Automatic Barcode Gap Discovery (ABGD) and Molecular Defined Operational Taxonomic Units (MOTU) methods, respectively, were performed for these and the 25 additional sequences of 10 species downloaded from GenBank. The results indicate that there are 34 species, 23 genera, and 6 families of the superfamily Acridoidea insects around the farmland, dam, and grassland of the Baiyangdian Wetland, including a new-record genus, Euchorthippus, and three new-record species, Euchorthippus unicolor, Atractomorpha psittacina and Oxya japonica. The DNA barcoding technology therefore is very efficient and helpful for identifying the species of the superfamily Acridoidea, although the morphological approach is still playing a key role in the species identifications.
白洋淀湿地蝗虫多样性调查及DNA条形码应用研究
DOI:10.17520/biods.2016331
[本文引用: 1]
为了解白洋淀湿地蝗虫的物种多样性及其分布情况, 探究DNA条形码技术在物种鉴定方面的可行性, 本研究对白洋淀湿地的蝗虫进行了采集调查。采用自行设计的cox1基因引物, 本研究共扩增得到21种蝗虫的cox1序列97条, 并利用构建NJ系统发育树法、计算遗传距离的ABGD法以及序列差异阈值分类的MOTU法对已得到的序列连同GenBank下载的10种25条cox1序列进行了比较分析。结果表明: 白洋淀湿地蝗总科昆虫共有6科23属34种, 包括1个新记录属, 即异爪蝗属(Euchorthippus), 和3个新记录种, 即素色异爪蝗(E. unicolor)、柳枝负蝗(Atractomorpha psittacina)和日本稻蝗(Oxya japonica); 蝗虫在白洋淀湿地主要分布在农田及其周围、堤坝及其周围以及淀边草丛; DNA条形码技术能够在种级阶元对蝗总科物种进行良好地解析, 但物种鉴定时, 不能完全脱离形态学方法。
DNA metabarcoding for biodiversity monitoring in a National Park: Screening for invasive and pest species
DOI:10.1111/1755-0998.13212 URL [本文引用: 1]
Barcoding animal life:Cytochrome c oxidase subunit
Assessment of current taxonomic assignment strategies for metabarcoding eukaryotes
DOI:10.1111/1755-0998.13407 URL [本文引用: 6]
Cryptic biodiversity in streams: A comparison of macroinvertebrate communities based on morphological and DNA barcode identifications
DOI:10.1086/675225 URL [本文引用: 1]
Reliable, verifiable and efficient monitoring of biodiversity via metabarcoding
DOI:10.1111/ele.12162
PMID:23910579
[本文引用: 1]
To manage and conserve biodiversity, one must know what is being lost, where, and why, as well as which remedies are likely to be most effective. Metabarcoding technology can characterise the species compositions of mass samples of eukaryotes or of environmental DNA. Here, we validate metabarcoding by testing it against three high-quality standard data sets that were collected in Malaysia (tropical), China (subtropical) and the United Kingdom (temperate) and that comprised 55,813 arthropod and bird specimens identified to species level with the expenditure of 2,505 person-hours of taxonomic expertise. The metabarcode and standard data sets exhibit statistically correlated alpha- and beta-diversities, and the two data sets produce similar policy conclusions for two conservation applications: restoration ecology and systematic conservation planning. Compared with standard biodiversity data sets, metabarcoded samples are taxonomically more comprehensive, many times quicker to produce, less reliant on taxonomic expertise and auditable by third parties, which is essential for dispute resolution. © 2013 The Authors. Ecology Letters published by John Wiley & Sons Ltd and CNRS.
Unearthing the potential of soil eDNA metabarcoding—Towards best practice advice for invertebrate biodiversity assessment
DOI:10.3389/fevo.2021.630560 URL [本文引用: 1]
Benthic eDNA metabarcoding provides accurate assessments of impact from oil extraction, and ecological insights
DOI:10.1016/j.ecolind.2021.108064 URL [本文引用: 1]
Rapid alignment-free phylogenetic identification of metagenomic sequences
DOI:10.1093/bioinformatics/btz068
PMID:30698645
[本文引用: 2]
Taxonomic classification is at the core of environmental DNA analysis. When a phylogenetic tree can be built as a prior hypothesis to such classification, phylogenetic placement (PP) provides the most informative type of classification because each query sequence is assigned to its putative origin in the tree. This is useful whenever precision is sought (e.g. in diagnostics). However, likelihood-based PP algorithms struggle to scale with the ever-increasing throughput of DNA sequencing.We have developed RAPPAS (Rapid Alignment-free Phylogenetic Placement via Ancestral Sequences) which uses an alignment-free approach, removing the hurdle of query sequence alignment as a preliminary step to PP. Our approach relies on the precomputation of a database of k-mers that may be present with non-negligible probability in relatives of the reference sequences. The placement is performed by inspecting the stored phylogenetic origins of the k-mers in the query, and their probabilities. The database can be reused for the analysis of several different metagenomes. Experiments show that the first implementation of RAPPAS is already faster than competing likelihood-based PP algorithms, while keeping similar accuracy for short reads. RAPPAS scales PP for the era of routine metagenomic diagnostics.Program and sources freely available for download at https://github.com/blinard-BIOINFO/RAPPAS.Supplementary data are available at Bioinformatics online.© The Author(s) 2019. Published by Oxford University Press. All rights reserved. For permissions, please e-mail: journals.permissions@oup.com.
Application of high throughput sequencing in metagenomics
高通量测序技术在宏基因组学中的应用
Benchmarking bioinformatic tools for fast and accurate eDNA metabarcoding species identification
DOI:10.1111/1755-0998.13430 URL [本文引用: 1]
IDTAXA: A novel approach for accurate taxonomic classification of microbiome sequences
DOI:10.1186/s40168-018-0521-5
PMID:30092815
[本文引用: 1]
Background: Microbiome studies often involve sequencing a marker gene to identify the microorganisms in samples of interest. Sequence classification is a critical component of this process, whereby sequences are assigned to a reference taxonomy containing known sequence representatives of many microbial groups. Previous studies have shown that existing classification programs often assign sequences to reference groups even if they belong to novel taxonomic groups that are absent from the reference taxonomy. This high rate of "over classification" is particularly detrimental in microbiome studies because reference taxonomies are far from comprehensive.Results: Here, we introduce IDTAXA, a novel approach to taxonomic classification that employs principles from machine learning to reduce over classification errors. Using multiple reference taxonomies, we demonstrate that IDTAXA has higher accuracy than popular classifiers such as BLAST, MAPSeq, QIIME, SINTAX, SPINGO, and the RDP Classifier. Similarly, IDTAXA yields far fewer over classifications on Illumina mock microbial community data when the expected taxa are absent from the training set. Furthermore, IDTAXA offers many practical advantages over other classifiers, such as maintaining low error rates across varying input sequence lengths and withholding classifications from input sequences composed of random nucleotides or repeats.Conclusions: IDTAXA's classifications may lead to different conclusions in microbiome studies because of the substantially reduced number of taxa that are incorrectly identified through over classification. Although misclassification error is relatively minor, we believe that many remaining misclassifications are likely caused by errors in the reference taxonomy. We describe how IDTAXA is able to identify many putative mislabeling errors in reference taxonomies, enabling training sets to be automatically corrected by eliminating spurious sequences. IDTAXA is part of the DECIPHER package for the R programming language, available through the Bioconductor repository or accessible online (http://DECIPHER.codes).
A DNA metabarcoding approach to characterize soil arthropod communities
DOI:10.1016/j.soilbio.2018.06.026 URL [本文引用: 1]
Thematic monitoring network of soil fauna diversity in China: Exploring the mystery of soils
DOI:10.17520/biods.2016019
[本文引用: 1]
The important roles of soil fauna diversity and associated indicative functions of environment changes have received increasing attention from both academic circles and government decision makers. This paper summarizes the current situation of soil fauna monitoring in developed countries and related work in China. We introduce the objectives and structure of the thematic monitoring network of soil fauna diversity (TMNSFD), and highlighted some aspects that need attention. The TMNSFD proposed to establish permanent monitoring plots within forest plots established by Chinese Forest Biodiversity Monitoring Network for monitoring soil fauna including earthworms, mites, springtails, nematodes and protists. During the years 2016-2020, TMNSFD may choose typical forest ecosystems as priority ecosystems for soil fauna monitoring, which cover temperate forest ecosystems (including broadleaved Korean pine mixed forests in Changbaishan, Jilin Province and warm temperate deciduous broadleaved forests in Donglingshan, Beijing), subtropical forest ecosystems (including typical subtropical evergreen broadleaved forests in Gutianshan, Zhejiang Province, lower subtropical evergreen broadleaved forests in Dinghushan, Guangdong Province, and north subtropical evergreen broad-leaved forests in Dujiangyan, Sichuan Province), tropical forest ecosystems (tropical rainforests in Xishuangbanna, Yunnan Province and Jianfengling, Hainan Province), as well as mountainous dark coniferous forests in Liziping, Sichuan Province. By 2030, TMNSFD soil fauna monitoring plots may cover various ecosystems including forests, grasslands, wetlands, deserts, farmland, urban areas and other typical ecosystems in different regions of China. TMNSFD emphasizes the value of applied molecular biology technology, unified monitoring methods, and manipulation experiments to simulate the effects of global change on soil fauna during the processes of monitoring. We propose monitoring soil fauna diversity once every 5 years in established monitoring plots. The objective of TMNSFD is to provide reliable and integrated data of soil fauna diversity via the establishment of standard monitoring methods and a data-sharing network at the national level, which could support the development of ecological civilization in China.
中国土壤动物多样性监测: 探知土壤中的奥秘
DOI:10.17520/biods.2016019
[本文引用: 1]
土壤动物多样性变化及其对环境的指示作用已被学术界和政府决策部门高度关注。本文从土壤动物多样性监测的重要性及面临的挑战、国内外土壤动物多样性监测概况等方面进行了评述, 提出了未来、尤其是2016-2020年我国土壤动物多样性监测的目标、站点布局、样地设置、监测类群和指标等, 并讨论了在制定土壤动物多样性监测方案时需考虑的问题, 有助于在全国开展多点化土壤动物多样性及分布状况的监测工作, 建立标准统一、数据共享的土壤动物监测网, 提供完整的、可信的监测数据, 为国家生态文明建设提供科技支撑。
BOLD: The barcode of life data system (http://www.barcodinglife.org)
Carrion fly-derived DNA metabarcoding is an effective tool for mammal surveys: Evidence from a known tropical mammal community
VSEARCH: A versatile open source tool for metagenomics
Space-time dynamics in monitoring neotropical fish communities using eDNA metabarcoding
DOI:10.1016/j.scitotenv.2020.142096 URL [本文引用: 1]
TaxonKit: A practical and efficient NCBI taxonomy toolkit
DOI:10.1016/j.jgg.2021.03.006
PMID:34001434
[本文引用: 1]
The National Center for Biotechnology Information (NCBI) Taxonomy is widely applied in biomedical and ecological studies. Typical demands include querying taxonomy identifier (TaxIds) by taxonomy names, querying complete taxonomic lineages by TaxIds, listing descendants of given TaxIds, and others. However, existed tools are either limited in functionalities or inefficient in terms of runtime. In this work, we present TaxonKit, a command-line toolkit for comprehensive and efficient manipulation of NCBI Taxonomy data. TaxonKit comprises seven core subcommands providing functions, including TaxIds querying, listing, filtering, lineage retrieving and reformatting, lowest common ancestor computation, and TaxIds change tracking. The practical functions, competitive processing performance, scalability with different scales of datasets and good accessibility could facilitate taxonomy data manipulations. TaxonKit provides free access under the permissive MIT license on GitHub, Brewsci, and Bioconda. The documents are also available at https://bioinf.shenwei.me/taxonkit/.Copyright © 2021 Institute of Genetics and Developmental Biology, Chinese Academy of Sciences, and Genetics Society of China. Published by Elsevier Ltd. All rights reserved.
Review of soil biodiversity research: History, current status and future challenges
土壤生物多样性研究: 历史、现状与挑战
Ecosystem biomonitoring with eDNA: Metabarcoding across the tree of life in a tropical marine environment
DOI:10.1038/s41598-017-12501-5
PMID:28947818
[本文引用: 1]
Effective marine management requires comprehensive data on the status of marine biodiversity. However, efficient methods that can document biodiversity in our oceans are currently lacking. Environmental DNA (eDNA) sourced from seawater offers a new avenue for investigating the biota in marine ecosystems. Here, we investigated the potential of eDNA to inform on the breadth of biodiversity present in a tropical marine environment. Directly sequencing eDNA from seawater using a shotgun approach resulted in only 0.34% of 22.3 million reads assigning to eukaryotes, highlighting the inefficiency of this method for assessing eukaryotic diversity. In contrast, using 'tree of life' (ToL) metabarcoding and 20-fold fewer sequencing reads, we could detect 287 families across the major divisions of eukaryotes. Our data also show that the best performing 'universal' PCR assay recovered only 44% of the eukaryotes identified across all assays, highlighting the need for multiple metabarcoding assays to catalogue biodiversity. Lastly, focusing on the fish genus Lethrinus, we recovered intra- and inter-specific haplotypes from seawater samples, illustrating that eDNA can be used to explore diversity beyond taxon identifications. Given the sensitivity and low cost of eDNA metabarcoding we advocate this approach be rapidly integrated into biomonitoring programs.
Towards next-generation biodiversity assessment using DNA metabarcoding
DOI:10.1111/j.1365-294X.2012.05470.x
PMID:22486824
[本文引用: 1]
Virtually all empirical ecological studies require species identification during data collection. DNA metabarcoding refers to the automated identification of multiple species from a single bulk sample containing entire organisms or from a single environmental sample containing degraded DNA (soil, water, faeces, etc.). It can be implemented for both modern and ancient environmental samples. The availability of next-generation sequencing platforms and the ecologists' need for high-throughput taxon identification have facilitated the emergence of DNA metabarcoding. The potential power of DNA metabarcoding as it is implemented today is limited mainly by its dependency on PCR and by the considerable investment needed to build comprehensive taxonomic reference libraries. Further developments associated with the impressive progress in DNA sequencing will eliminate the currently required DNA amplification step, and comprehensive taxonomic reference libraries composed of whole organellar genomes and repetitive ribosomal nuclear DNA can be built based on the well-curated DNA extract collections maintained by standardized barcoding initiatives. The near-term future of DNA metabarcoding has an enormous potential to boost data acquisition in biodiversity research.© 2012 Blackwell Publishing Ltd.
Towards an integrative understanding of soil biodiversity
DOI:10.1111/brv.12567
[本文引用: 1]
Soil is one of the most biodiverse terrestrial habitats. Yet, we lack an integrative conceptual framework for understanding the patterns and mechanisms driving soil biodiversity. One of the underlying reasons for our poor understanding of soil biodiversity patterns relates to whether key biodiversity theories (historically developed for aboveground and aquatic organisms) are applicable to patterns of soil biodiversity. Here, we present a systematic literature review to investigate whether and how key biodiversity theories (species-energy relationship, theory of island biogeography, metacommunity theory, niche theory and neutral theory) can explain observed patterns of soil biodiversity. We then discuss two spatial compartments nested within soil at which biodiversity theories can be applied to acknowledge the scale-dependent nature of soil biodiversity.
Multiomic approach to analyze infant gut microbiota: Experimental and analytical method optimization
DOI:10.3390/biom11070999 URL [本文引用: 1]
Testing multiple substrates for terrestrial biodiversity monitoring using environmental DNA metabarcoding
DOI:10.1111/1755-0998.13148 URL [本文引用: 1]
Sorting specimen-rich invertebrate samples with cost-effective NGS barcodes: Validating a reverse workflow for specimen processing
DOI:10.1111/1755-0998.12751
PMID:29314756
[本文引用: 1]
Biologists frequently sort specimen-rich samples to species. This process is daunting when based on morphology, and disadvantageous if performed using molecular methods that destroy vouchers (e.g., metabarcoding). An alternative is barcoding every specimen in a bulk sample and then presorting the specimens using DNA barcodes, thus mitigating downstream morphological work on presorted units. Such a "reverse workflow" is too expensive using Sanger sequencing, but we here demonstrate that is feasible with an next-generation sequencing (NGS) barcoding pipeline that allows for cost-effective high-throughput generation of short specimen-specific barcodes (313 bp of COI; laboratory cost <$0.50 per specimen) through next-generation sequencing of tagged amplicons. We applied our approach to a large sample of tropical ants, obtaining barcodes for 3,290 of 4,032 specimens (82%). NGS barcodes and their corresponding specimens were then sorted into molecular operational taxonomic units (mOTUs) based on objective clustering and Automated Barcode Gap Discovery (ABGD). High diversity of 88-90 mOTUs (4% clustering) was found and morphologically validated based on preserved vouchers. The mOTUs were overwhelmingly in agreement with morphospecies (match ratio 0.95 at 4% clustering). Because of lack of coverage in existing barcode databases, only 18 could be accurately identified to named species, but our study yielded new barcodes for 48 species, including 28 that are potentially new to science. With its low cost and technical simplicity, the NGS barcoding pipeline can be implemented by a large range of laboratories. It accelerates invertebrate species discovery, facilitates downstream taxonomic work, helps with building comprehensive barcode databases and yields precise abundance information.© 2018 John Wiley & Sons Ltd.
Testing three pipelines for 18S rDNA-based metabarcoding of soil faunal diversity
DOI:10.1007/s11427-012-4423-7 URL [本文引用: 3]
Applications and prospects of metabarcoding in environmental monitoring of phytoplankton community
DNA宏条形码(metabarcoding)技术在浮游植物群落监测研究中的应用

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}