



除了少数形成肉眼可见子实体的大型真菌(主要是担子菌门和少数子囊菌门的类群)外, 微生物主要指肉眼难见的微型生物, 包括细菌、真菌、古菌、病毒等类群, 是地球上出现时间最早、分布最广泛、个体数量最多、物种和基因多样性十分丰富的生物类群。然而在自然界中, 由于绝大多数微生物难以在野外形成肉眼可见的子实体和在人工培养基上生长, 只能通过传统的分离培养方法, 并结合形态学、生理生化和DNA序列分析进行物种鉴定。因此传统方法难以全面监测环境中微生物的多样性, 限制了人们对微生物多样性及其生态功能的认知。随着分子生物学技术, 特别是第二代DNA测序技术和生物信息学的发展, 微生物多样性的研究得到极大的推动(图1)。本文将对近年来的微生物多样性分布格局与维持、群落构建以及功能属性多样性研究的进展进行综述。
图1
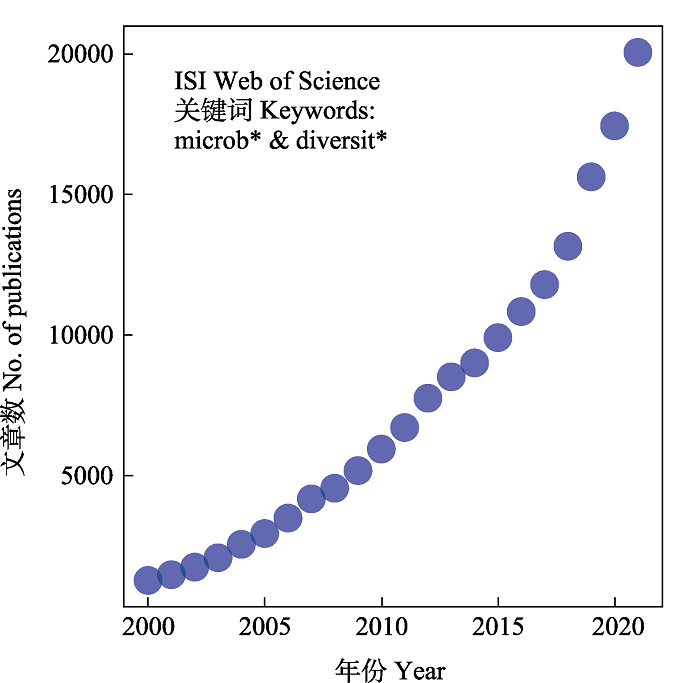
图1
微生物多样性研究发表文章数量逐年增加。在ISI Web of Science数据库中以microb*和diversit*为关键词进行检索, 检索期限为2000-2020, 检索时间为2022年9月15日。每个点代表每年发表的文章数。
Fig. 1
Numbers of published papers of microbial diversity in recent years. We performed search in ISI Web of Science using keywords microb* and diversit*, ranged from 2000 to 2020, at the date of September 15, 2022.
1 微生物多样性
微生物具有丰富的物种多样性。据推算, 全球微生物多样性高达1万亿种(Locey & Lennon, 2016)。保守估计地球上病毒和噬菌体的个体数量在1031 (Dion et al, 2020), 物种多样性在1亿种以上(Rohwer, 2003)。例如, Gregory等(2019)从全球186个海洋样品中检测到了195,728个病毒种群, Camarillo-Guerrero等(2021)从28,060个人类肠道宏基因组中检测到了142,809个噬菌体种群。在已知的27个古菌门中, 仅有6个门的古菌有可培养的菌株(Baker et al, 2020), GTDB数据库中收录的拥有基因组的古菌物种数量为3,412种, 仅占已知基因组原核生物物种数量的5% (
真菌是生物多样性最丰富的真核生物类群之一。早期根据英国学者基于已知的真菌物种数与高等植物物种数的比例(6:1)推算全球有150万种真菌(Hawksworth, 1991)。由于该项研究没有考虑其他气候带(如热带等)的情况, 大家普遍认为这个数据低估了真菌的多样性。目前学界较普遍接受的保守估计认为全球约有220-380万种真菌(Hawksworth & Lücking, 2017)。近期基于扩增子数据的Chao-1多样性估测方式预测全球真菌多样性为620多万种(Baldrian et al, 2022)。目前, 全世界只报道了约14.7万种真菌(
1.1 微生物多样性分布格局
微生物是否存在与动植物相似的多样性纬度分布格局, 即从赤道向两极多样性逐渐降低(Hillebrand, 2004)? Delgado-Baquerizo等(2016)对全球600多份土壤的荟萃分析(meta-analysis)表明在南半球土壤细菌群落多样性随纬度升高而递减, 而在北半球没有显著纬度分布规律, 而全球189个土壤样品的宏基因组分析发现, 细菌多样性在中纬度的温带地区最高, 向赤道和极地地区逐渐降低(Bahram et al, 2018)。单独对南半球647个地点的研究发现土壤古菌多样性从赤道到南极逐渐降低(Delgado-Baquerizo et al, 2018), 单独对北半球中高纬度地区深海的研究发现古菌多样性随着纬度升高而升高(Danovaro et al, 2016)。Thurber (2009)对全球68个地点10年的研究发现噬菌体多样性存在着从赤道向极地递减的规律, 可能是由于噬菌体多样性与其宿主微生物的多样性和组成有关(Rohwer, 2003)。多项研究均表明真菌多样性从赤道到两极随着纬度的增加而降低(Tedersoo et al, 2014; Bahram et al, 2018), 而对全球3,084份土壤样品ITS扩增子数据荟萃分析的结果则发现真菌多样性在高纬度地区最高(Větrovský et al, 2019)。对真菌功能类群的研究发现腐生真菌、病原真菌、丛枝菌根真菌、植物内生真菌的多样性也随着纬度的增加而降低(Arnold & Lutzoni, 2007; Tedersoo et al, 2014; Davison et al, 2015)。然而, 外生菌根真菌多样性则在中高纬度地区最高, 向赤道和极地地区逐渐降低(Tedersoo & Nara, 2010; Gao et al, 2013; Tedersoo et al, 2014)。酵母型真菌多样性则在中纬度的温带地区最低, 向赤道和两极逐渐增加(Tedersoo et al, 2014)。总之, 微生物多样性纬度分布格局在不同类群和不同研究中的结论不尽相同。
1.2 微生物多样性驱动因子
微生物多样性受到各种生物和非生物因子的影响。例如, 大量的研究发现细菌多样性与土壤pH (Fierer & Jackson, 2006)、海拔(Bryant et al, 2008)、温度(Zhou et al, 2016)、土壤有机质含量(Tian et al, 2018)、干旱(Xu et al, 2018)等密切相关。古菌多样性受到土壤pH值、水分、有效氮、有机磷、有机质等影响(Leff et al, 2015; Zhang et al, 2019; Jiao et al, 2021; Wang et al, 2022)。真菌多样性受到植物多样性、生产力、系统发育、地形、土壤养分和水分等影响(Tedersoo et al, 2014; Ji et al, 2019; Zheng et al, 2021)。例如, 对我国从南到北的12个森林大样地的研究发现土壤真菌多样性与植物多样性和土壤养分相关(Ji et al, 2019)。而对浙江古田山24 ha大样地的研究发现真菌多样性在山脊生境受植物多样性、土壤养分和湿度影响, 而在山谷生境受到样方凹凸度的影响(Gao et al, 2017)。此外, 在长白山的研究发现随着海拔的增加, 植物的多样性减少, 但真菌的多样性没有显著的变化(Shen et al, 2014)。
1.3 人类活动对微生物多样性的影响
人类世是指人类活动显著改变地质结构的时代, 地球最晚在1950年以后全面进入人类世(Waters et al, 2016)。人类世的标志包括城市化、工业化、农业集约化、化石燃料的大量开采、人造材料的生产、碳氮磷等循环的显著改变、海平面上升、生物入侵和生物灭绝等(Waters et al, 2016)。研究发现, 施肥降低了细菌和真菌的多样性, 干旱降低了细菌的多样性却增加了真菌的多样性, 而且作用强度依赖于生态系统类型和干扰程度(Maestre et al, 2015; Liu et al, 2020; Zhou et al, 2020; Xu et al, 2021)。增温影响细菌多样性的升高或降低(Sheik et al, 2011; Wu et al, 2022), 但对真菌多样性影响不大(Zhou et al, 2020)。全球25个草地氮磷添加实验的整合分析表明, 施氮磷增加了病原真菌相对多度, 而降低了共生真菌的相对多度(Lekberg et al, 2021)。对温带淡水浮游群落的研究表明, 气候变暖促进了病毒种群多样性动态的时间变化(Frenken et al, 2020)。对上海城市土壤的研究发现距城市中心越远, 细菌和真菌多样性越高(Liu et al, 2022)。对美国堪萨斯州栎树叶际真菌的研究也发现城市地区叶际真菌多样性低于郊区(Jumpponen & Jones, 2009)。相反, 加拿大蒙特利尔7种树木的研究表明城市密度越高, 叶表生细菌多样性越高(Laforest-Lapointe et al, 2017)。总之, 目前关于人类世对微生物多样性的影响尚缺乏全面系统的评估(Berg & Cernava, 2022)。
2 微生物群落构建
驱动生物群落构建的诸多因素可归纳为4个过程: 选择(selection)、扩散(dispersal)、成种/多样化/突变(speciation/diversification/mutation)、生态漂变(ecological drift) (Vellend, 2010, 2016)。前人综述归纳了这4个过程对细菌(Nemergut et al, 2013; Petro et al, 2017; Fitzpatrick et al, 2020)、真菌(Peay et al, 2016)、病原菌(Seabloom et al, 2015)、菌根真菌(高程和郭良栋, 2013; Bogar & Peay, 2017)等群落构建的影响。此前对微生物群落构建的研究通常聚焦于环境选择和扩散限制, 而忽略生物互作、生态漂变、均匀扩散、多样化等过程。本文尝试更全面的综述4个过程在微生物群落构建中的贡献。
2.1 选择过程
选择过程包含环境选择和生物互作两个层面的内容。
环境选择。大量前人研究发现微生物群落结构与各种生物和非生物因子及人类活动相关。例如, 细菌和真菌及其功能类群组成受到空间、土壤、植被、气候等因素的影响(Gao et al, 2015; Vályi et al, 2016; van der Linde et al, 2018; Yang et al, 2019)。古菌群落组成与土壤水分、养分和pH值等因素密切相关(Jiao et al, 2019; Li et al, 2021; Wang et al, 2021; Zhang et al, 2021)。病毒组成则与海拔、土壤pH值和钙含量等相关(Adriaenssens et al, 2017)。根据作用方式的不同, 环境选择对微生物群落的影响不同。例如, 异质性选择(heterogeneous selection)过程会导致微生物群落间组成差异变大, 而同质性选择(homogenous selection)过程则会导致微生物群落间组成趋同(Stegen et al, 2015; Huang et al, 2020; Jiao et al, 2020; Fodelianakis et al, 2022)。不管异质性选择还是同质性选择, 通常认为环境选择对微生物群落的影响是确定性的过程(Zhou & Ning, 2017)。
生物互作。对于微生物组数据, 目前主要利用共现网络表征生物之间的互作关系(Coyte et al, 2015; Neilson et al, 2017; de Vries et al, 2018; Wang et al, 2018)。然而, 共现网络在多大程度上对应现实中的生物互作仍存在争论(Banerjee et al, 2018, 2019; Röttjers & Faust, 2019)。胁迫梯度假说认为随着胁迫增加, 生物间的竞争将减弱而互惠增强(Bertness & Callaway, 1994; Callaway et al, 2002)。对微生物的研究发现, 资源限制、互养(cross-feeding)、重金属胁迫等都可促进微生物间的合作(Hoek et al, 2016; Velez et al, 2018; Hammarlund & Harcombe, 2019; Piccardi et al, 2019)。然而, 关于生物互作对微生物群落构建的影响, 目前在理论和分析方法(网络分析、代谢互补分析、毒素-抗毒素互作分析、合成群落等)上都仍处于快速发展阶段。
2.2 生态漂变过程
生态漂变是指物种相对多度的随机波动(Gilbert & Levine, 2017)。通常认为环境选择压力比较小的时候生态漂变对群落的影响较大, 导致随机性升高。研究发现当干旱、盐碱、pH、养分限制和捕食者等的胁迫消除后, 细菌、真菌、丛枝菌根真菌等群落相异性显著升高(Chase, 2007, 2009, 2010; Zhou et al, 2014; Gao et al, 2016; Zhang et al, 2016; Tripathi et al, 2018)。此外, 另一个假说认为当群落较小的时候生态漂变对群落的影响较大, 导致随机性升高(Vellend et al, 2014)。近期对植物生长发育过程中真菌组的研究发现随机性强度与真菌群落大小限制负相关, 表明生态漂变在植物生长发育早期导致真菌群落相异性升高(Gao et al, 2020)。然而, 目前大多数关于微生物群落的研究通常将随机性等同于生态漂变。
2.3 扩散过程
扩散是指生物个体在空间的迁移(Vellend, 2010)。扩散过程对群落的影响与其强度有关。扩散限制通常会导致群落相异性升高, 而均匀扩散则造成群落相异性降低(Stegen et al, 2015)。目前, 对微生物扩散的直接检测还非常少(Peay et al, 2012; Adams et al, 2013; Chaudhary et al, 2020; Paz et al, 2021), 大多数研究利用微生物的分布来推测扩散过程(Nemergut et al, 2013; Fitzpatrick et al, 2020)。扩散限制对微生物群落的影响通常与研究的尺度密切相关: 在区域和全球尺度上扩散限制作用较大, 而在局域和微小尺度上较小。相比扩散限制, 均匀扩散对微生物群落的影响还缺乏研究。
2.4 成种/多样化/突变过程
3 微生物功能性状多样性
功能性状(functional trait)分析最早应用于植物生态学, 是指对植物定植、存活、生长和死亡存在显著潜在影响的一系列植物属性, 能够单独或联合指示生态系统对环境变化的响应, 而且能够对生态系统过程产生强烈影响, 现广泛应用于植物种群、群落和生态系统生态学(刘晓娟和马克平, 2015)。相比于植物, 微生物虽然个体小、形态简单, 但是其定植能力高、活力强、功能丰富, 对环境变化的响应快, 因此微生物功能性状与微生物的多样性分布、群落构建、物质循环以及环境变化等过程密切相关。然而, 目前关于微生物功能性状的研究才刚刚起步, 还缺乏统一的理论框架。例如, 不同的框架分别将微生物功能性状划分为: (1)定性性状和定量性状; (2)基因组性状和表型性状; (3)响应性状和效应性状; (4) r策略与k策略性状; (5)寡营养-富营养策略性状; (6)竞争-胁迫耐受-杂生(C-S-R)策略性状 (7)高产-资源获取-胁迫耐受(Y-A-S)策略性状等。
3.1 细菌功能性状
环境宏基因组技术的发展大大促进了细菌和古菌功能多样性的研究(Westoby et al, 2021; Yang, 2021)。由于个体微小和形态简单, 对细菌和古菌功能性状的研究主要依赖于其基因组信息, 如rRNA基因拷贝数、B族维生素合成、GC含量、基因数量、基因组大小、革兰氏阳性/阴性、产孢基因、毒素-抗毒素系统等(Kearns & Shade, 2018; Guittar et al, 2019)。大量研究发现rRNA基因拷贝数随着细菌群落演替而下降, 表明早期具有高rRNA基因拷贝数的采取r策略的细菌类群被后期具有较低rRNA基因拷贝数且采取k策略的细菌类群所替代(Kim et al, 2016; Nemergut et al, 2016; Ortiz-Álvarez et al, 2018; Prest et al, 2018)。然而, 关于宏基因组性状与适应策略的关系仍缺乏共识。例如, Krause等(2014)提出高的rRNA基因拷贝数与r策略有关, 而Fierer (2017)认为高的rRNA拷贝数与k策略有关。宏基因组中蕴含的大量信息有助于理解生态系统物质循环和生物互作等过程。例如, Nelson等(2016)对116个地点的宏基因组分析发现氮循环基因的分布主要受到生境类型、土壤碳和氮含量等的影响。由此可见, 功能性状可以反映细菌和古菌的分布格局、群落演替以及生态系统中的物质循环等过程。
3.2 真菌功能性状
前人尝试从不同角度对真菌功能性状进行了总结。例如, Aguilar-Trigueros等(2014)总结了与腐生和共生生活策略相关的真菌性状, 提出共生性状包括真菌避免或克服植物免疫系统胁迫以及与资源获取和双向转运相关的性状, 腐生性状则包括有机大分子分解酶的产生和分泌以及与其他微生物进行资源竞争并避免土壤动物捕食等性状。Treseder和Lennon (2015)总结真菌驱动生态系统动态的功能性状, 包括木质纤维分解、氮和磷元素转化以及与胁迫耐受等相关的功能性状。Bahram和Netherway (2021)则将真菌功能性状划分为生态生理性状、繁殖性状、扩散性状、基因组性状等。Nguyen等(2016)建立了FUNGuilds数据库, 根据分类信息将真菌划分为不同功能类群。Zanne等(2020)建立了FunFun数据库, 将80多项真菌性状划分为7大类: 生殖分配、生殖、水和无机养分获取、碳分配、营养分配、个体大小、碳和有机营养获取等。Põlme等(2020)整合FUNGuilds和FunFun数据库而构建了FungalTraits数据库, 包含了10,210个真菌属, 涵盖17个与生活类型相关的性状。然而, 目前关于真菌功能性状存在大量关键类群数据缺失、主观定性数据为主、普适理论和框架缺失等问题。
菌根(mycorrhiza)是土壤真菌与植物根系形成的互惠共生体, 普遍存在于绝大多数的陆地植物根际(Smith & Read, 2008)。在菌根共生体系中, 宿主植物为菌根真菌提供生长所需的碳水化合物, 而菌根真菌帮助植物从环境中吸收氮磷等无机养分和水分, 提高植物对环境胁迫和病原菌的抵抗能力, 拓展宿主植物的现实生态位(Smith & Read, 2008)。此外, 菌根真菌形成的地下菌丝网络能将同种或不同种植物个体连接起来, 影响营养物质和能量在这些个体间的分配, 从而调控植物个体间的相互作用(He et al, 2003; Booth, 2004; Wipf et al, 2019; van’t Padje et al, 2021)。根据真菌与植物根系形成的解剖结构以及植物类群, 将菌根分为7种类型: 外生菌根(ectomycorrhiza)、丛枝菌根(arbuscular mycorrhiza)、内外生菌根(ectendomycorrhiza)、杜鹃菌根(ericoid mycorrhiza)、浆果鹃类菌根(arbutoid mycorrhiza)、水晶兰类菌根(monotropoid mycorrhiza)、兰科菌根(orchid mycorrhiza) (Smith & Read, 2008)。其中, 丛枝菌根和外生菌根是自然界中分布最广泛的菌根类型, 大量的研究发现丛枝菌根和外生菌根植物在植物-土壤反馈(Bennett et al, 2017)、负密度制约(Chen et al, 2019)、植物营养经济学谱(Averill et al, 2019)、土壤碳库动态(Craig et al, 2018)等方面存在巨大差异, 进而影响生态系统功能和植物群落构建(Liu et al, 2012; Bagchi et al, 2014)。植物菌根类型的准确鉴定是探究其介导的生态过程的前提(Brundrett & Tedersoo, 2019; Bueno et al, 2019)。Soudzilovskaia等(2020)构建了FungalRoot数据库, 为陆地植物菌根类型的快速鉴定提供了便利。
丛枝菌根。丛枝菌根真菌可与80%的陆地植物形成共生关系, 其中丛枝(arbuscule)、根外菌丝(extra-hyphae)和孢子(spore)是丛枝菌根最典型的3类形态结构: 丛枝是真菌-植物进行资源交换的界面, 根外菌丝大大拓展了植物根系对土壤养分的吸收范围, 而休眠孢子则在耐受外界不良环境和扩散等方面具有重要作用(Smith & Read, 2008)。前人研究发现丛枝菌根真菌的丛枝、根外菌丝和孢子等受到植物、土壤、气候等因素的影响(Han et al, 2020; Babalola et al, 2022)。菌根真菌的功能性状很大程度上由上述3种形态结构所决定。例如, Chagnon等(2013)根据Grime (1974)的模型提出: 竞争型丛枝菌根真菌(如巨孢囊霉科)将大量植物来源的碳分配到根外菌丝以增强对土壤养分的吸收; 杂生型丛枝菌根真菌生长迅速, 在生态系统重建过程中快速定植植物根系并完成产孢; 胁迫耐受型丛枝菌根真菌则具有最高的碳利用效率, 菌丝生长缓慢。Chaudhary等(2020)发现丛枝菌根真菌在空气中的扩散主要受到孢子大小影响。Deveautour等(2020)发现丛枝菌根真菌孢子的黑色素含量随着干旱胁迫程度升高而增加。Gao等(2022)在干旱农田的研究发现长期灌溉造成耐旱丛枝菌根真菌丧失, 而演替早期的r策略丛枝菌根真菌通过植物根系分泌的独脚金内酯加速共生关系的建立, 而演替后期的c策略丛枝菌根真菌通过脂几丁质寡糖调控共生关系的形成。
外生菌根。外生菌根是真菌(保守估计约25,000种)与宿主植物(约8,500种)形成的互惠共生体, 其宿主植物包括温带、亚热带和热带森林中的建群种和优势树种, 如壳斗科、桦木科、松科、杨柳科和龙脑香科等植物(Brundrett & Tedersoo, 2018)。根据根外菌丝的有无和长度, 外生菌根真菌可被划分为接触型(contact)、短距离(short distance)、中距离(medium distance)和长距离(long distance)等不同探测类型(exploration type) (Agerer, 2001)。前人研究发现外生菌根真菌的探测类型与植物根系密度、植物生产力、土壤湿度、CO2浓度等密切相关(Bakker et al, 2006; Koide et al, 2008; Kranabetter et al, 2009; Fransson, 2012; Chen et al, 2018)。然而, van der Linde等(2018)对欧洲外生菌根的研究发现超过30%的外生菌根真菌物种具有形态上的塑性, 表明仅从分类鉴定信息无法准确推测外生菌根真菌的探测类型。综上所述, 虽然微生物具有丰富的多样性和生态功能, 但是微生物功能性状研究刚刚起步, 需要加强功能性状在微生物多样性分布、群落构建、物质循环和能量流动过程中的作用, 及其对全球环境变化响应的研究。
4 展望
目前, 国内外学者开展了大量的微生物多样性研究, 特别是在微生物多样性分布格局与维持、群落构建机制以及生态功能等方面取得了瞩目的进展。但是相比于动植物, 微生物多样性研究还存在较大的差距, 例如我们对不同微生物类群的物种和功能基因多样性缺乏深入了解。针对微生物多样性研究现状, 提出未来的微生物多样性研究的重点领域(图2):
图2

图2
微生物多样性进展和展望概念图
Fig. 2
Concept framework of progress and perspective of microbial diversity
(1)环境宏真菌组学研究。宏基因组、宏转录组等组学技术已被广泛应用于环境微生物组的物种和功能基因多样性研究。虽然自然界中真菌具有丰富的物种和功能多样性, 但是由于环境中(如土壤等)的原核微生物(细菌、古菌等)的个体数(细胞数)远高于真核微生物(真菌), 提取的环境总DNA或mRNA由原核微生物占主导, 导致了环境宏基因组测序结果中超过90%的DNA序列来源于原核微生物, 只有不到10%的DNA序列来源于真菌, 因此, 现有的宏基因组学技术难以全面监测环境中真菌组的物种和功能基因多样性。为了深入阐明环境真菌组的功能多样性, 未来需将宏基因组学、真核宏转录组学、靶向探针富集, 以及新的高通量测序等技术综合应用于环境真菌组学研究。
(2)微生物多样性与生态系统功能关系研究。生物多样性与生态系统功能(BEF)关系是生态学的重要研究领域。目前, 主要开展了植物多样性与生态系统功能关系研究, 发现植物多样性与生产力存在正相关性; 此外, 已逐步从物种多样性发展到功能多样性和beta多样性, 从单一功能到生态系统多功能性。然而, 我们对微生物多样性与生态系统功能的关系知之甚少, 因此, 未来需深入开展微生物多样性与生态系统多功能性的关系研究。
(3)微生物互作网络的生态功能研究。自然界中任何生物都不是独立存在的, 生物之间存在着广泛的相互作用, 如共生、共存、寄生、对抗等。不同的生物互作涉及到信号识别与交流、资源竞争与分配、物理和化学对抗、基因流、群感效应等复杂过程。生态网络分析已应用于微生物类群间以及微生物与动植物的互作关系研究。然而, 这种基于相关性的分析是否能真实反映生物间的相互作用还存在争议。另外, 微生物生态网络分析只是分析生物类群间的互作关系, 并不能解释微生物生态功能。因此, 未来的微生物生态网络分析要整合物种、系统演化关系、功能性状、功能基因等信息, 深入阐明群落的物种共存、稳定性等生态功能。
致谢
感谢李杏春、姚慧、王聪、陈沛霖、刘娜娜、仰剑霞、于清弋、朱晓华、史加勉、朱欢欢、刘欣等在文献资料收集方面提供的帮助。
参考文献
Dispersal in microbes: Fungi in indoor air are dominated by outdoor air and show dispersal limitation at short distances
DOI:10.1038/ismej.2013.28 URL [本文引用: 1]
Environmental drivers of viral community composition in Antarctic soils identified by viromics
DOI:10.1186/s40168-017-0301-7
PMID:28724405
[本文引用: 1]

The Antarctic continent is considered the coldest and driest place on earth with simple ecosystems, devoid of higher plants. Soils in the ice-free regions of Antarctica are known to harbor a wide range of microorganisms from primary producers to grazers, yet their ecology and particularly the role of viruses is poorly understood. In this study, we examined the virus community structures of 14 soil samples from the Mackay Glacier region.Viral communities were extracted from soil and the dsDNA was extracted, amplified using single-primer amplification, and sequenced using the Ion Torrent Proton platform. Metadata on soil physico-chemistry was collected from all sites. Both read and contig datasets were analyzed with reference-independent and reference-dependent methods to assess viral community structures and the influence of environmental parameters on their distribution.We observed a high heterogeneity in virus signatures, independent of geographical proximity. Tailed bacteriophages were dominant in all samples, but the incidences of the affiliated families Siphoviridae and Myoviridae were inversely correlated, suggesting direct competition for hosts. Viruses of the families Phycodnaviridae and Mimiviridae were present at significant levels in high-diversity soil samples and were found to co-occur, implying little competition between them. Combinations of soil factors, including pH, calcium content, and site altitude, were found to be the main drivers of viral community structure.The pattern of viral community structure with higher levels of diversity at lower altitude and pH, and co-occurring viral families, suggests that these cold desert soil viruses interact with each other, the host, and the environment in an intricate manner, playing a potentially crucial role in maintaining host diversity and functioning of the microbial ecosystem in the extreme environments of Antarctic soil.
Exploration types of ectomycorrhizae
DOI:10.1007/s005720100108 URL [本文引用: 1]
Ecological understanding of root-infecting fungi using trait-based approaches
DOI:10.1016/j.tplants.2014.02.006
PMID:24613596
[本文引用: 1]
Classification schemes have been popular to tame the diversity of root-infecting fungi. However, the usefulness of these schemes is limited to descriptive purposes. We propose that a shift to a multidimensional trait-based approach to disentangle the saprotrophic-symbiotic continuum will provide a better framework to understand fungal evolutionary ecology. Trait information reflecting the separation of root-infecting fungi from free-living soil relatives will help to understand the evolutionary process of symbiosis, the role that species interactions play in maintaining their large diversity in soil and in planta, and their contributions at the ecosystem level. Methodological advances in several areas such as microscopy, plant immunology, and metatranscriptomics represent emerging opportunities to populate trait databases. Copyright © 2014 Elsevier Ltd. All rights reserved.
Diversity and host range of foliar fungal endophytes: Are tropical leaves biodiversity hotspots?
Fungal endophytes are found in asymptomatic photosynthetic tissues of all major lineages of land plants. The ubiquity of these cryptic symbionts is clear, but the scale of their diversity, host range, and geographic distributions are unknown. To explore the putative hyperdiversity of tropical leaf endophytes, we compared endophyte communities along a broad latitudinal gradient from the Canadian arctic to the lowland tropical forest of central Panama. Here, we use molecular sequence data from 1403 endophyte strains to show that endophytes increase in incidence, diversity, and host breadth from arctic to tropical sites. Endophyte communities from higher latitudes are characterized by relatively few species from many different classes of Ascomycota, whereas tropical endophyte assemblages are dominated by a small number of classes with a very large number of endophytic species. The most easily cultivated endophytes from tropical plants have wide host ranges, but communities are dominated by a large number of rare species whose host range is unclear. Even when only the most easily cultured species are considered, leaves of tropical trees represent hotspots of fungal species diversity, containing numerous species not yet recovered from other biomes. The challenge remains to recover and identify those elusive and rarely cultured taxa with narrower host ranges, and to elucidate the ecological roles of these little-known symbionts in tropical forests.
Global imprint of mycorrhizal fungi on whole-plant nutrient economics
Nitrogen fertilisation disrupts the temporal dynamics of arbuscular mycorrhizal fungal hyphae but not spore density and community composition in a wheat field
DOI:10.1111/nph.18043
PMID:35179789
[本文引用: 1]
Elucidating the temporal dynamics of arbuscular mycorrhizal (AM) fungi is critical for understanding their functions. Furthermore, research investigating the temporal dynamics of AM fungi in response to agricultural practices remain in its infancy. We investigated the effect of nitrogen fertilization and watering reduction on the temporal dynamics of AM fungi, across the lifespan of wheat. Nitrogen fertilization decreased AM fungal spore density, extra-radical hyphal density, and intra-radical colonization rate in both watering conditions. Nitrogen fertilization affected AM fungal community composition in soil but not in roots, regardless of watering conditions. The temporal analysis revealed that AM fungal extra-radical hyphal density and intra-radical colonization rate were higher under conventional watering and lower under reduced watering in March than in other growth stages at low (≤ 70 kg N ha yr ) but not at high (≥ 140) nitrogen fertilization levels. AM fungal spore density was lower in June than in other growth stages and community composition varied with plant development at all nitrogen fertilization levels, regardless of watering conditions. This study demonstrates that high nitrogen fertilization levels disrupt the temporal dynamics of AM fungal hyphal growth but not sporulation and community composition.This article is protected by copyright. All rights reserved.
Pathogens and insect herbivores drive rainforest plant diversity and composition
DOI:10.1038/nature12911 URL [本文引用: 1]
Structure and function of the global topsoil microbiome
DOI:10.1038/s41586-018-0386-6 URL [本文引用: 2]
Fungi as mediators linking organisms and ecosystems
DOI:10.1093/femsre/fuab058 URL [本文引用: 1]
Diversity, ecology and evolution of archaea
DOI:10.1038/s41564-020-0715-z
PMID:32367054
[本文引用: 1]
Compared to bacteria, our knowledge of archaeal biology is limited. Historically, microbiologists have mostly relied on culturing and single-gene diversity surveys to understand Archaea in nature. However, only six of the 27 currently proposed archaeal phyla have cultured representatives. Advances in genomic sequencing and computational approaches are revolutionizing our understanding of Archaea. The recovery of genomes belonging to uncultured groups from the environment has resulted in the description of several new phyla, many of which are globally distributed and are among the predominant organisms on the planet. In this Review, we discuss how these genomes, together with long-term enrichment studies and elegant in situ measurements, are providing insights into the metabolic capabilities of the Archaea. We also debate how such studies reveal how important Archaea are in mediating an array of ecological processes, including global carbon and nutrient cycles, and how this increase in archaeal diversity has expanded our view of the tree of life and early archaeal evolution, and has provided new insights into the origin of eukaryotes.
Fine root distribution of trees and understory in mature stands of maritime pine (Pinus pinaster) on dry and humid sites
DOI:10.1007/s11104-006-9024-4 URL [本文引用: 1]
High-throughput sequencing view on the magnitude of global fungal diversity
DOI:10.1007/s13225-021-00472-y URL [本文引用: 1]
Keystone taxa as drivers of microbiome structure and functioning
DOI:10.1038/s41579-018-0024-1
PMID:29789680
[本文引用: 1]
Microorganisms have a pivotal role in the functioning of ecosystems. Recent studies have shown that microbial communities harbour keystone taxa, which drive community composition and function irrespective of their abundance. In this Opinion article, we propose a definition of keystone taxa in microbial ecology and summarize over 200 microbial keystone taxa that have been identified in soil, plant and marine ecosystems, as well as in the human microbiome. We explore the importance of keystone taxa and keystone guilds for microbiome structure and functioning and discuss the factors that determine their distribution and activities.
Reply to ‘Can We Predict Microbial Keystones?’
DOI:10.1038/s41579-018-0133-x PMID:30538306 [本文引用: 1]
Plant-soil feedbacks and mycorrhizal type influence temperate forest population dynamics
DOI:10.1126/science.aai8212
PMID:28082590
[本文引用: 1]
Feedback with soil biota is an important determinant of terrestrial plant diversity. However, the factors regulating plant-soil feedback, which varies from positive to negative among plant species, remain uncertain. In a large-scale study involving 55 species and 550 populations of North American trees, the type of mycorrhizal association explained much of the variation in plant-soil feedbacks. In soil collected beneath conspecifics, arbuscular mycorrhizal trees experienced negative feedback, whereas ectomycorrhizal trees displayed positive feedback. Additionally, arbuscular mycorrhizal trees exhibited strong conspecific inhibition at multiple spatial scales, whereas ectomycorrhizal trees exhibited conspecific facilitation locally and less severe conspecific inhibition regionally. These results suggest that mycorrhizal type, through effects on plant-soil feedbacks, could be an important contributor to population regulation and community structure in temperate forests.Copyright © 2017, American Association for the Advancement of Science.
The plant microbiota signature of the Anthropocene as a challenge for microbiome research
DOI:10.1186/s40168-021-01224-5
PMID:35346369
[本文引用: 1]
One promise of the recently presented microbiome definition suggested that, in combination with unifying concepts and standards, microbiome research could be important for solving new challenges associated with anthropogenic-driven changes in various microbiota. With this commentary we want to further elaborate this suggestion, because we noticed specific signatures in microbiota affected by the Anthropocene.Here, we discuss this based on a review of available literature and our own research targeting exemplarily the plant microbiome. It is not only crucial for plants themselves but also linked to planetary health. We suggest that different human activities are commonly linked to a shift of diversity and evenness of the plant microbiota, which is also characterized by a decrease of host specificity, and an increase of r-strategic microbes, pathogens, and hypermutators. The resistome, anchored in the microbiome, follows this shift by an increase of specific antimicrobial resistance (AMR) mechanisms as well as an increase of plasmid-associated resistance genes. This typical microbiome signature of the Anthropocene is often associated with dysbiosis and loss of resilience, and leads to frequent pathogen outbreaks. Although several of these observations are already confirmed by meta-studies, this issue requires more attention in upcoming microbiome studies.Our commentary aims to inspire holistic studies for the development of solutions to restore and save microbial diversity for ecosystem functioning as well as the closely connected planetary health. Video abstract.© 2022. The Author(s).
Positive interactions in communities
DOI:10.1016/0169-5347(94)90088-4
PMID:21236818
[本文引用: 1]
Current concepts of the role of interspecific interactions in communities have been shaped by a profusion of experimental studies of interspecific competition over the past few decades. Evidence for the importance of positive interactions - facilitations - in community organization and dynamics has accrued to the point where it warrants formal inclusion into community ecology theory, as it has been in evolutionary biology.Copyright © 1994. Published by Elsevier Ltd.
Mycorrhizal networks mediate overstorey- understorey competition in a temperate forest
DOI:10.1111/j.1461-0248.2004.00605.x URL [本文引用: 1]
Misdiagnosis of mycorrhizas and inappropriate recycling of data can lead to false conclusions
DOI:10.1111/nph.15440
PMID:30191568
[本文引用: 1]
We draw attention to a worrying trend for the uncritical use of 'recycled' mycorrhizal data to compile host species lists that include obvious errors or undertake risky analyses that correlate mycorrhizal colonisation levels with environmental or physiological factors despite inherent limitations in datasets. We are not suggesting that all meta-studies are wrong, only that more care should be taken to resolve what can safely be done with recycled mycorrhizal data in the future. We also recommend that mycorrhizal species lists should be checked against standard references since the majority of EM hosts and NM plant belong to families that are well resolved. However, additional research is required in cases where plant families have multiple root types within genera or occur in habitats where mycorrhizal associations are often suppressed (see Brundrett & Tedersoo, 2018). We hope that the mycorrhizal science community will work together more closely in the future to develop and enforce standards for mycorrhizal diagnosis and to share carefully corrected datasets for realistic meta-studies.© 2018 The Authors. New Phytologist © 2018 New Phytologist Trust.
Evolutionary history of mycorrhizal symbioses and global host plant diversity
DOI:10.1111/nph.14976
PMID:29355963
[本文引用: 1]
Contents Summary 1108 I. Introduction 1108 II. Mycorrhizal plant diversity at global and local scales 1108 III. Mycorrhizal evolution in plants: a brief update 1111 IV. Conclusions and perspectives 1114 References 1114 SUMMARY: The majority of vascular plants are mycorrhizal: 72% are arbuscular mycorrhizal (AM), 2.0% are ectomycorrhizal (EcM), 1.5% are ericoid mycorrhizal and 10% are orchid mycorrhizal. Just 8% are completely nonmycorrhizal (NM), whereas 7% have inconsistent NM-AM associations. Most NM and NM-AM plants are nutritional specialists (e.g. carnivores and parasites) or habitat specialists (e.g. hydrophytes and epiphytes). Mycorrhizal associations are consistent in most families, but there are exceptions with complex roots (e.g. both EcM and AM). We recognize three waves of mycorrhizal evolution, starting with AM in early land plants, continuing in the Cretaceous with multiple new NM or EcM linages, ericoid and orchid mycorrhizas. The third wave, which is recent and ongoing, has resulted in root complexity linked to rapid plant diversification in biodiversity hotspots.© 2018 The Authors. New Phytologist © 2018 New Phytologist Trust.
Microbes on mountainsides: Contrasting elevational patterns of bacterial and plant diversity
Misdiagnosis and uncritical use of plant mycorrhizal data are not the only elephants in the room
DOI:10.1111/nph.15976 PMID:31246312 [本文引用: 1]
Positive interactions among alpine plants increase with stress
DOI:10.1038/nature00812 URL [本文引用: 1]
Massive expansion of human gut bacteriophage diversity
DOI:10.1016/j.cell.2021.01.029
PMID:33606979
[本文引用: 1]
Bacteriophages drive evolutionary change in bacterial communities by creating gene flow networks that fuel ecological adaptions. However, the extent of viral diversity and its prevalence in the human gut remains largely unknown. Here, we introduce the Gut Phage Database, a collection of ∼142,000 non-redundant viral genomes (>10 kb) obtained by mining a dataset of 28,060 globally distributed human gut metagenomes and 2,898 reference genomes of cultured gut bacteria. Host assignment revealed that viral diversity is highest in the Firmicutes phyla and that ∼36% of viral clusters (VCs) are not restricted to a single species, creating gene flow networks across phylogenetically distinct bacterial species. Epidemiological analysis uncovered 280 globally distributed VCs found in at least 5 continents and a highly prevalent phage clade with features reminiscent of p-crAssphage. This high-quality, large-scale catalog of phage genomes will improve future virome studies and enable ecological and evolutionary analysis of human gut bacteriophages.Copyright © 2021 The Authors. Published by Elsevier Inc. All rights reserved.
A trait-based framework to understand life history of mycorrhizal fungi
DOI:10.1016/j.tplants.2013.05.001 URL [本文引用: 1]
Drought mediates the importance of stochastic community assembly
Stochastic community asssembly causes higher biodiversity in more productive environments
DOI:10.1126/science.1187820 URL [本文引用: 1]
Predators temper the relative importance of stochastic processes in the assembly of prey metacommunities
DOI:10.1111/j.1461-0248.2009.01362.x
PMID:19723282
[本文引用: 1]
Communities assemble through a combination of stochastic processes, which can make environmentally similar communities divergent (high beta-diversity), and deterministic processes, which can make environmentally similar communities convergent (low beta-diversity). Top predators can influence both stochasticity (e.g. colonization and extinction events) and determinism (e.g. size of the realized species pool), in community assembly, and thus their net effect is unknown. We investigated how predatory fish influenced the scaling of prey diversity in ponds at local and regional spatial scales. While fish reduced both local and regional richness, their effects were markedly more intense at the regional scale. Underlying this result was that the presence of fish made localities within metacommunities more similar in their community composition (lower beta-diversity), suggesting that fish enhance the deterministic, relative to the stochastic, components of community assembly. Thus, the presence of predators can alter fundamental mechanisms of community assembly and the scaling of diversity within metacommunities.
Trait-based aerial dispersal of arbuscular mycorrhizal fungi
DOI:10.1111/nph.16667 URL [本文引用: 2]
Differential soil fungus accumulation and density dependence of trees in a subtropical forest
DOI:10.1126/science.aau1361
PMID:31604314
[本文引用: 1]
The mechanisms underlying interspecific variation in conspecific negative density dependence (CNDD) are poorly understood. Using a multilevel modeling approach, we combined long-term seedling demographic data from a subtropical forest plot with soil fungal community data by means of DNA sequencing to address the feedback of various guilds of soil fungi on the density dependence of trees. We show that mycorrhizal type mediates tree neighborhood interactions at the community level, and much of the interspecific variation in CNDD is explained by how tree species differ in their fungal density accumulation rates as they grow. Species with higher accumulation rates of pathogenic fungi suffered more from CNDD, whereas species with lower CNDD had higher accumulation rates of ectomycorrhizal fungi, suggesting that mutualistic and pathogenic fungi play important but opposing roles.Copyright © 2019 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. No claim to original U.S. Government Works.
Root diameter predicts the extramatrical hyphal exploration distance of the ectomycorrhizal fungal community
The ecology of the microbiome: Networks, competition, and stability
DOI:10.1126/science.aad2602
PMID:26542567
[本文引用: 1]
The human gut harbors a large and complex community of beneficial microbes that remain stable over long periods. This stability is considered critical for good health but is poorly understood. Here we develop a body of ecological theory to help us understand microbiome stability. Although cooperating networks of microbes can be efficient, we find that they are often unstable. Counterintuitively, this finding indicates that hosts can benefit from microbial competition when this competition dampens cooperative networks and increases stability. More generally, stability is promoted by limiting positive feedbacks and weakening ecological interactions. We have analyzed host mechanisms for maintaining stability-including immune suppression, spatial structuring, and feeding of community members-and support our key predictions with recent data. Copyright © 2015, American Association for the Advancement of Science.
Tree mycorrhizal type predicts within-site variability in the storage and distribution of soil organic matter
DOI:10.1111/gcb.14132
PMID:29573504
[本文引用: 1]
Forest soils store large amounts of carbon (C) and nitrogen (N), yet how predicted shifts in forest composition will impact long-term C and N persistence remains poorly understood. A recent hypothesis predicts that soils under trees associated with arbuscular mycorrhizas (AM) store less C than soils dominated by trees associated with ectomycorrhizas (ECM), due to slower decomposition in ECM-dominated forests. However, an incipient hypothesis predicts that systems with rapid decomposition-e.g. most AM-dominated forests-enhance soil organic matter (SOM) stabilization by accelerating the production of microbial residues. To address these contrasting predictions, we quantified soil C and N to 1 m depth across gradients of ECM-dominance in three temperate forests. By focusing on sites where AM- and ECM-plants co-occur, our analysis controls for climatic factors that covary with mycorrhizal dominance across broad scales. We found that while ECM stands contain more SOM in topsoil, AM stands contain more SOM when subsoil to 1 m depth is included. Biomarkers and soil fractionations reveal that these patterns are driven by an accumulation of microbial residues in AM-dominated soils. Collectively, our results support emerging theory on SOM formation, demonstrate the importance of subsurface soils in mediating plant effects on soil C and N, and indicate that shifts in the mycorrhizal composition of temperate forests may alter the stabilization of SOM.© 2018 John Wiley & Sons Ltd.
Macroecological drivers of archaea and bacteria in benthic deep-sea ecosystems
DOI:10.1126/sciadv.1500961 URL [本文引用: 1]
Global assessment of arbuscular mycorrhizal fungus diversity reveals very low endemism
DOI:10.1126/science.aab1161
PMID:26315436
[本文引用: 1]
The global biogeography of microorganisms remains largely unknown, in contrast to the well-studied diversity patterns of macroorganisms. We used arbuscular mycorrhizal (AM) fungus DNA from 1014 plant-root samples collected worldwide to determine the global distribution of these plant symbionts. We found that AM fungal communities reflected local environmental conditions and the spatial distance between sites. However, despite AM fungi apparently possessing limited dispersal ability, we found 93% of taxa on multiple continents and 34% on all six continents surveyed. This contrasts with the high spatial turnover of other fungal taxa and with the endemism displayed by plants at the global scale. We suggest that the biogeography of AM fungi is driven by unexpectedly efficient dispersal, probably via both abiotic and biotic vectors, including humans. Copyright © 2015, American Association for the Advancement of Science.
Soil bacterial networks are less stable under drought than fungal networks
DOI:10.1038/s41467-018-05516-7
PMID:30072764
[本文引用: 1]
Soil microbial communities play a crucial role in ecosystem functioning, but it is unknown how co-occurrence networks within these communities respond to disturbances such as climate extremes. This represents an important knowledge gap because changes in microbial networks could have implications for their functioning and vulnerability to future disturbances. Here, we show in grassland mesocosms that drought promotes destabilising properties in soil bacterial, but not fungal, co-occurrence networks, and that changes in bacterial communities link more strongly to soil functioning during recovery than do changes in fungal communities. Moreover, we reveal that drought has a prolonged effect on bacterial communities and their co-occurrence networks via changes in vegetation composition and resultant reductions in soil moisture. Our results provide new insight in the mechanisms through which drought alters soil microbial communities with potential long-term consequences, including future plant community composition and the ability of aboveground and belowground communities to withstand future disturbances.
Carbon content and climate variability drive global soil bacterial diversity patterns
DOI:10.1002/ecm.1216 URL [本文引用: 1]
Ecological drivers of soil microbial diversity and soil biological networks in the Southern Hemisphere
DOI:10.1002/ecy.2137
PMID:29315530
[本文引用: 1]
The ecological drivers of soil biodiversity in the Southern Hemisphere remain underexplored. Here, in a continental survey comprising 647 sites, across 58 degrees of latitude between tropical Australia and Antarctica, we evaluated the major ecological patterns in soil biodiversity and relative abundance of ecological clusters within a co-occurrence network of soil bacteria, archaea and eukaryotes. Six major ecological clusters (modules) of co-occurring soil taxa were identified. These clusters exhibited strong shifts in their relative abundances with increasing distance from the equator. Temperature was the major environmental driver of the relative abundance of ecological clusters when Australia and Antarctica are analyzed together. Temperature, aridity, soil properties and vegetation types were the major drivers of the relative abundance of different ecological clusters within Australia. Our data supports significant reductions in the diversity of bacteria, archaea and eukaryotes in Antarctica vs. Australia linked to strong reductions in temperature. However, we only detected small latitudinal variations in soil biodiversity within Australia. Different environmental drivers regulate the diversity of soil archaea (temperature and soil carbon), bacteria (aridity, vegetation attributes and pH) and eukaryotes (vegetation type and soil carbon) across Australia. Together, our findings provide new insights into the mechanisms driving soil biodiversity in the Southern Hemisphere.© 2018 by the Ecological Society of America.
Biogeography of arbuscular mycorrhizal fungal spore traits along an aridity gradient, and responses to experimental rainfall manipulation
DOI:10.1016/j.funeco.2019.100899 URL [本文引用: 1]
Phage diversity, genomics and phylogeny
DOI:10.1038/s41579-019-0311-5
PMID:32015529
[本文引用: 1]
Recent advances in viral metagenomics have enabled the rapid discovery of an unprecedented catalogue of phages in numerous environments, from the human gut to the deep ocean. Although these advances have expanded our understanding of phage genomic diversity, they also revealed that we have only scratched the surface in the discovery of novel viruses. Yet, despite the remarkable diversity of phages at the nucleotide sequence level, the structural proteins that form viral particles show strong similarities and conservation. Phages are uniquely interconnected from an evolutionary perspective and undergo multiple events of genetic exchange in response to the selective pressure of their hosts, which drives their diversity. In this Review, we explore phage diversity at the structural, genomic and community levels as well as the complex evolutionary relationships between phages, moulded by the mosaicity of their genomes.
Embracing the unknown: Disentangling the complexities of the soil microbiome
DOI:10.1038/nrmicro.2017.87
PMID:28824177
[本文引用: 1]
Soil microorganisms are clearly a key component of both natural and managed ecosystems. Despite the challenges of surviving in soil, a gram of soil can contain thousands of individual microbial taxa, including viruses and members of all three domains of life. Recent advances in marker gene, genomic and metagenomic analyses have greatly expanded our ability to characterize the soil microbiome and identify the factors that shape soil microbial communities across space and time. However, although most soil microorganisms remain undescribed, we can begin to categorize soil microorganisms on the basis of their ecological strategies. This is an approach that should prove fruitful for leveraging genomic information to predict the functional attributes of individual taxa. The field is now poised to identify how we can manipulate and manage the soil microbiome to increase soil fertility, improve crop production and improve our understanding of how terrestrial ecosystems will respond to environmental change.
The diversity and biogeography of soil bacterial communities
The plant microbiome: From ecology to reductionism and beyond
DOI:10.1146/annurev-micro-022620-014327
PMID:32530732
[本文引用: 2]
Methodological advances over the past two decades have propelled plant microbiome research, allowing the field to comprehensively test ideas proposed over a century ago and generate many new hypotheses. Studying the distribution of microbial taxa and genes across plant habitats has revealed the importance of various ecological and evolutionary forces shaping plant microbiota. In particular, selection imposed by plant habitats strongly shapes the diversity and composition of microbiota and leads to microbial adaptation associated with navigating the plant immune system and utilizing plant-derived resources. Reductionist approaches have demonstrated that the interaction between plant immunity and the plant microbiome is, in fact, bidirectional and that plants, microbiota, and the environment shape a complex chemical dialogue that collectively orchestrates the plantmicrobiome. The next stage in plant microbiome research will require the integration of ecological and reductionist approaches to establish a general understanding of the assembly and function in both natural and managed environments.
Microdiversity characterizes prevalent phylogenetic clades in the glacier-fed stream microbiome
DOI:10.1038/s41396-021-01106-6 URL [本文引用: 1]
Elevated CO2 impacts ectomycorrhiza- mediated forest soil carbon flow: Fungal biomass production, respiration and exudation
DOI:10.1016/j.funeco.2011.10.001 URL [本文引用: 1]
Warming advances virus population dynamics in a temperate freshwater plankton community
Successional adaptive strategies revealed by correlating arbuscular mycorrhizal fungal abundance with host plant gene expression
Distribution pattern and maintenance of ectomycorrhizal fungus diversity
DOI:10.3724/SP.J.1003.2013.11055
[本文引用: 2]
Ectomycorrhiza (ECM) are symbionts formed between soil fungi and plant root systems, in which the fungus exchanges soil-derived nutrients for carbohydrates obtained from the host plant. As an important component of terrestrial ecosystems, ECM fungi can play an essential role in biodiversity maintenance and plant community succession. Understanding the distribution pattern and maintenance of ECM fungal diversity is therefore critical to the study of biodiversity and ecosystem functioning. An analysis of results of recent research indicates that ECM fungal diversity increases with increasing latitude, i.e. from tropical to subtropical and temperate regions. The role of dispersal in ECM fungal distribution is dependent on spatial scale. Thus, it has been found to be weak across global and local scales, but strong at regional and small scales. At the local scale, its influence has also been shown to be host-dominant dependent; thus, it is important in host non-dominant ecosystems, but not in host dominant ecosystems. Selection by plant, animal, microbe and abiotic factors can also affect the distribution pattern of ECM fungi, according to studies of temperate ecosystems. In contrast, studies of tropical ecosystems indicate that selection on ECM fungal distribution can be either strong or weak. ECM fungal diversity is also influenced by plant diversity and productivity. The plant diversity hypothesis at host genus-level fits well with ECM fungal diversity in temperate, subtropical and tropical forest ecosystems; in contrast, the productivity diversity hypothesis is only supported by some studies in temperate forest ecosystems. We propose that future studies should focus on the distribution pattern, maintenance mechanism and ecosystem function of ECM fungal diversity at a global scale, taking account of scenarios of global climate change.
外生菌根真菌多样性的分布格局与维持机制研究进展
DOI:10.3724/SP.J.1003.2013.11055
[本文引用: 2]
外生菌根(ectomycorrhiza, ECM)是由土壤真菌与陆地植物根系形成的一种互惠共生体。ECM真菌从寄主植物中获取生长所需的碳源, 同时促进寄主吸收氮、磷等矿物营养物质和水分。作为生态系统的重要组分, ECM真菌在生态系统的演替和多样性维持中发挥着重要的作用, 因而揭示ECM真菌多样性的分布格局与维持机制是生物多样性与生态系统功能研究的热点领域之一。本文对ECM真菌多样性的最新研究进展进行了综合分析, 相关研究显示, 从热带到亚热带、温带森林, 每种寄主植物上ECM真菌的平均物种数逐渐升高。扩散和选择过程都影响ECM真菌的分布格局, 其中扩散对ECM真菌分布的影响具有空间尺度依赖性, 即在全球和局域尺度上, 扩散对ECM真菌分布的影响较弱, 而在区域和小尺度上很强。同时, 在局域尺度上, 扩散对ECM真菌的分布具有寄主植物优势度依赖性, 即在寄主植物不占优势的生态系统中, 扩散对ECM真菌的分布有明显作用; 而在寄主植物占优势的生态系统中则无影响。植物、动物、微生物和非生物因素的选择也都影响ECM真菌的分布格局, 其中在温带地区所有研究均表明选择对ECM真菌的分布有影响, 但是在热带地区有的研究表明选择对ECM真菌的分布有影响, 而有的研究则显示无影响。植物的多样性和生产力都能影响ECM真菌的多样性, 其中在温带、亚热带和热带森林中寄主植物属的多样性决定ECM真菌多样性, 而植物生产力多样性假说只在一些温带的研究中得到证实。未来的研究重点应关注全球尺度, 特别是在全球气候变化背景下的ECM真菌多样性的分布格局、维持机制及其生态系统功能等方面。
Increased precipitation, rather than warming, exerts a strong influence on arbuscular mycorrhizal fungal community in a semiarid steppe ecosystem
DOI:10.1139/cjb-2015-0210 URL [本文引用: 1]
Fungal community assembly in drought-stressed Sorghum shows stochasticity, selection, and universal ecological dynamics
DOI:10.1038/s41467-019-13913-9 URL [本文引用: 1]
Relationships between soil fungal and woody plant assemblages differ between ridge and valley habitats in a subtropical mountain forest
DOI:10.1111/nph.14287
PMID:28164340
[本文引用: 1]
Elucidating interactions of above-ground and below-ground communities in different habitat types is essential for understanding biodiversity maintenance and ecosystem functioning. Using 454 pyrosequencing of ITS2 sequences we examined the relationship between subtropical mountain forest soil fungal communities, abiotic conditions, and plant communities using correlation and partial models. Ridge and valley habitats with differing fungal communities were delineated. Total, saprotrophic and pathogenic fungal richness were significantly correlated with plant species richness and/or soil nutrients and moisture in the ridge habitat, but with habitat convexity or basal area of Castanopsis eyrei in the valley habitat. Ectomycorrhizal (EM) fungal richness was significantly correlated with basal area of C. eyrei and total EM plants in the ridge and valley habitats, respectively. Total, saprotrophic, pathogenic and EM fungal compositions were significantly correlated with plant species composition and geographic distance in the ridge habitat, but with various combinations of plant species composition, plant species richness, soil C : N ratio and pH or no variables in the valley habitat. Our findings suggest that mechanisms influencing soil fungal diversity and community composition differ between ridge and valley habitats, and relationships between fungal and woody plant assemblages depend on habitat types in the subtropical forest ecosystem.© 2016 The Authors. New Phytologist © 2016 New Phytologist Trust.
Host plant genus-level diversity is the best predictor of ectomycorrhizal fungal diversity in a Chinese subtropical forest
PMID:24624421
Microbial diversity is generally far higher than plant diversity, but the relationship between microbial diversity and plant diversity remains enigmatic. To shed light on this problem, we examined the diversity of a key guild of root-associated microbes,that is, ectomycorrhizal (EM) fungi along a plant diversity gradient in a Chinese subtropical forest. The results indicated that EM fungal diversity was positively correlated with host plant diversity. Furthermore, this relationship was best predicted by host genus-level diversity, rather than species-level diversity or family-level diversity. The generality of this finding was extended beyond our study system through the analyses of 100 additional studies of EM fungal communities from tropical and temperate forests.Here as well, EM fungal lineage composition was significantly affected by EM plant diversity levels, and some EM fungal lineages were co-associated with some host plant genera. These results suggest a general diversity maintenance mechanism for host-specific microbes based on higher order host plant phylogenetic diversity.
Community assembly of ectomycorrhizal fungi along a subtropical secondary forest succession
DOI:10.1111/nph.13068
PMID:25303438
[本文引用: 1]
Environmental selection and dispersal limitation are two of the primary processes structuring biotic communities in ecosystems, but little is known about these processes in shaping soil microbial communities during secondary forest succession. We examined the communities of ectomycorrhizal (EM) fungi in young, intermediate and old forests in a Chinese subtropical ecosystem, using 454 pyrosequencing. The EM fungal community consisted of 393 operational taxonomic units (OTUs), belonging to 21 EM fungal lineages, in which three EM fungal lineages and 11 EM fungal OTUs showed significantly biased occurrence among the young, intermediate and old forests. The EM fungal community was structured by environmental selection and dispersal limitation in old forest, but only by environmental selection in young, intermediate, and whole forests. Furthermore, the EM fungal community was affected by different factors in the different forest successional stages, and the importance of these factors in structuring EM fungal community dramatically decreased along the secondary forest succession series. This study suggests that different assembly mechanisms operate on the EM fungal community at different stages in secondary subtropical forest succession. © 2014 The Authors New Phytologist © 2014 New Phytologist Trust.
Unique and common traits in mycorrhizal symbioses
DOI:10.1038/s41579-020-0402-3 URL [本文引用: 1]
Ecological drift and the distribution of species diversity
Marine DNA viral macro- and microdiversity from pole to pole
DOI:S0092-8674(19)30341-1
PMID:31031001
[本文引用: 1]
Microbes drive most ecosystems and are modulated by viruses that impact their lifespan, gene flow, and metabolic outputs. However, ecosystem-level impacts of viral community diversity remain difficult to assess due to classification issues and few reference genomes. Here, we establish an ∼12-fold expanded global ocean DNA virome dataset of 195,728 viral populations, now including the Arctic Ocean, and validate that these populations form discrete genotypic clusters. Meta-community analyses revealed five ecological zones throughout the global ocean, including two distinct Arctic regions. Across the zones, local and global patterns and drivers in viral community diversity were established for both macrodiversity (inter-population diversity) and microdiversity (intra-population genetic variation). These patterns sometimes, but not always, paralleled those from macro-organisms and revealed temperate and tropical surface waters and the Arctic as biodiversity hotspots and mechanistic hypotheses to explain them. Such further understanding of ocean viruses is critical for broader inclusion in ecosystem models.Copyright © 2019 Elsevier Inc. All rights reserved.
Vegetation classification by reference to strategies
DOI:10.1038/250026a0 URL [本文引用: 1]
Trait-based community assembly and succession of the infant gut microbiome
DOI:10.1038/s41467-019-08377-w
PMID:30710083
[本文引用: 1]
The human gut microbiome develops over early childhood and aids in food digestion and immunomodulation, but the mechanisms driving its development remain elusive. Here we use data curated from literature and online repositories to examine trait-based patterns of gut microbiome succession in 56 infants over their first three years of life. We also develop a new phylogeny-based approach of inferring trait values that can extend readily to other microbial systems and questions. Trait-based patterns suggest that infant gut succession begins with a functionally variable cohort of taxa, adept at proliferating rapidly within hosts, which gradually matures into a more functionally uniform cohort of taxa adapted to thrive in the anoxic gut and disperse between anoxic patches as oxygen-tolerant spores. Trait-based composition stabilizes after the first year, while taxonomic turnover continues unabated, suggesting functional redundancy in the traits examined. Trait-based approaches powerfully complement taxonomy-based approaches to understanding the mechanisms of microbial community assembly and succession.
Refining the stress gradient hypothesis in a microbial community
Responses of arbuscular mycorrhizal fungi to nitrogen addition: A meta-analysis
DOI:10.1111/gcb.15369 URL [本文引用: 1]
The fungal dimension of biodiversity: Magnitude, significance, and conservation
DOI:10.1016/S0953-7562(09)80810-1 URL [本文引用: 1]
Fungal diversity revisited: 2.2 to 3.8 million species
Nitrogen transfer within and between plants through common mycorrhizal networks (CMNs)
DOI:10.1080/713608315 URL [本文引用: 1]
On the generality of the latitudinal diversity gradient
DOI:10.1086/381004 URL [本文引用: 1]
Resource availability modulates the cooperative and competitive nature of a microbial cross-feeding mutualism
DOI:10.1371/journal.pbio.1002540 URL [本文引用: 1]
Microecological Koch’s postulates reveal that intestinal microbiota dysbiosis contributes to shrimp white feces syndrome
DOI:10.1186/s40168-020-00802-3 URL [本文引用: 1]
Late Quaternary climate change explains soil fungal community composition rather than fungal richness in forest ecosystems
DOI:10.1002/ece3.5247 URL [本文引用: 2]
Linking phylogenetic niche conservatism to soil archaeal biogeography, community assembly and species coexistence
DOI:10.1111/geb.13313 URL [本文引用: 1]
Environmental filtering drives distinct continental atlases of soil archaea between dryland and wetland agricultural ecosystems
DOI:10.1186/s40168-019-0630-9
PMID:30709414
[本文引用: 1]
Understanding the spatial distributions and ecological diversity of soil archaeal communities in agricultural ecosystems is crucial for improvements in crop productivity. Here, we conducted a comprehensive, continental-scale survey of soil archaeal communities in adjacent pairs of maize (dryland) and rice (wetland) fields in eastern China.We revealed the consequential roles of environmental filtering in driving archaeal community assembly for both maize and rice fields. Rice fields, abundant with Euryarchaeota, had higher archaeal diversity and steeper distance-decay slopes than maize fields dominated by Thaumarchaeota. Dominant soil archaea showed distinct continental atlases and niche differentiation between dryland and wetland habitats, where they were associated with soil pH and mean annual temperature, respectively. After identifying their environmental preferences, we grouped the dominant archaeal taxa into different ecological clusters and determined the unique co-occurrence patterns within each cluster. Using this empirical dataset, we built a continental atlas of soil archaeal communities to provide reliable estimates of their spatial distributions in agricultural ecosystems.Environmental filtering plays a crucial role in driving the distinct continental atlases of dominant soil archaeal communities between dryland and wetland, with contrasting strategies of archaeal-driven nutrient cycling within these two agricultural ecosystems. These findings improve our ability to predict how soil archaeal communities respond to environmental changes and to manage soil archaeal communities for provisioning of agricultural ecosystem services.
Balance between community assembly processes mediates species coexistence in agricultural soil microbiomes across Eastern China
DOI:10.1038/s41396-019-0522-9 URL [本文引用: 1]
Massively parallel 454 sequencing indicates hyperdiverse fungal communities in temperate Quercus macrocarpa phyllosphere
DOI:10.1111/j.1469-8137.2009.02990.x
PMID:19674337
[本文引用: 1]
* This study targeted the fungal communities in the phyllosphere of Quercus macrocarpa and compared the fungal species richness, diversity and community composition among trees located within and outside a small urban center using recently developed 454 sequencing and DNA tagging. * The results indicate that the fungal phyllosphere communities are extremely diverse and strongly dominated by ascomycetes, with Microsphaeropsis [two Operational Taxonomic Units (OTUs); 23.6%], Alternaria (six OTUs; 16.1%), Epicoccum (one OTU; 6.0%) and Erysiphe (two OTUs; 5.9%) as the most abundant genera. * Although the sequencing effort averaged 1000 reads per tree and detected nearly 700 distinct molecular OTUs at 95% internal transcribed spacer 1 similarity, the richness of the hyperdiverse phyllosphere communities could not be reliably estimated as nearly one-half of the molecular OTUs were singletons. * The fungal communities within and outside the urban center differed in richness and diversity, which were lower within the urban development. The two land-use types contained communities that were distinct and more than 10% of the molecular OTUs differed in their frequency.
Trait-based patterns of microbial dynamics in dormancy potential and heterotrophic strategy: Case studies of resource-based and post-press succession
DOI:10.1038/s41396-018-0194-x URL [本文引用: 1]
Shifts in bacterial community structure during succession in a glacier foreland of the High Arctic
DOI:10.1093/femsec/fiw213 URL [本文引用: 1]
Ectomycorrhizal fungi and the biotrophy-saprotrophy continuum
DOI:10.1111/j.1469-8137.2008.02401.x PMID:18312537 [本文引用: 1]
Diversity and species distribution of ectomycorrhizal fungi along productivity gradients of a southern boreal forest
DOI:10.1007/s00572-008-0208-z
PMID:18941804
[本文引用: 1]
Coniferous forests with diverse ectomycorrhizal fungus (EMF) communities are associated with nutrient-poor, acidic soils but there is some debate whether EMF can be equally adapted to more productive, nitrogen-rich sites. We compared EMF species distribution and diversity along a replicated productivity gradient in a southern boreal forest of British Columbia (Canada). Roots from subalpine fir (Abies lasiocarpa) saplings of the understory were sampled and EMF species were identified by morphotypes supplemented with ITS rDNA analysis. There were significant changes in the distribution and abundance of 74 EMF species along the productivity gradient, with as little as 24% community similarity among contrasting sites. Species richness per plot increased asymptotically with foliar nitrogen concentrations of subalpine fir, demonstrating that many EMF species were well suited to soils with high rates of nitrogen mineralization. EMF species abundance in relation to site productivity included parabolic, negative linear, and positive exponential curves. Both multi-site and more narrowly distributed EMF were documented, and a diverse mix of mantle exploration types was present across the entire productivity gradient. The results demonstrate strong associations of EMF fungal species with edaphic characteristics, especially nitrogen availability, and a specialization in EMF communities that may contribute to the successful exploitation of such contrasting extremes in soil fertility by a single tree host.
Trait-based approaches for understanding microbial biodiversity and ecosystem functioning
DOI:10.3389/fmicb.2014.00251
PMID:24904563
[本文引用: 1]
In ecology, biodiversity-ecosystem functioning (BEE) research has seen a shift in perspective from taxonomy to function in the last two decades, with successful application of trait-based approaches. This shift offers opportunities for a deeper mechanistic understanding of the role of biodiversity in maintaining multiple ecosystem processes and services. In this paper, we highlight studies that have focused on BEE of microbial communities with an emphasis on integrating trait-based approaches to microbial ecology. In doing so, we explore some of the inherent challenges and opportunities of understanding BEE using microbial systems. For example, microbial biologists characterize communities using gene phylogenies that are often unable to resolve functional traits. Additionally, experimental designs of existing microbial BEE studies are often inadequate to unravel BEE relationships. We argue that combining eco-physiological studies with contemporary molecular tools in a trait-based framework can reinforce our ability to link microbial diversity to ecosystem processes. We conclude that such trait-based approaches are a promising framework to increase the understanding of microbial BEE relationships and thus generating systematic principles in microbial ecology and more generally ecology.
Tree leaf bacterial community structure and diversity differ along a gradient of urban intensity
Consistent responses of soil microbial communities to elevated nutrient inputs in grasslands across the globe
Nitrogen and phosphorus fertilization consistently favor pathogenic over mutualistic fungi in grassland soils
DOI:10.1038/s41467-021-23605-y
PMID:34108462
[本文引用: 1]
Ecosystems across the globe receive elevated inputs of nutrients, but the consequences of this for soil fungal guilds that mediate key ecosystem functions remain unclear. We find that nitrogen and phosphorus addition to 25 grasslands distributed across four continents promotes the relative abundance of fungal pathogens, suppresses mutualists, but does not affect saprotrophs. Structural equation models suggest that responses are often indirect and primarily mediated by nutrient-induced shifts in plant communities. Nutrient addition also reduces co-occurrences within and among fungal guilds, which could have important consequences for belowground interactions. Focusing only on plots that received no nutrient addition, soil properties influence pathogen abundance globally, whereas plant community characteristics influence mutualists, and climate influence saprotrophs. We show consistent, guild-level responses that enhance our ability to predict shifts in soil function related to anthropogenic eutrophication, which can have longer-term consequences for plant communities.
Coexistence patterns of soil methanogens are closely tied to methane generation and community assembly in rice paddies
DOI:10.1186/s40168-020-00978-8
PMID:33482926
[本文引用: 1]
Soil methanogens participate in complex interactions, which determine the community structures and functions. Studies continue to seek the coexistence patterns of soil methanogens, influencing factors and the contribution to methane (CH) production, which are regulated primarily by species interactions, and the functional significance of these interactions. Here, methane emissions were measured in rice paddies across the Asian continent, and the complex interactions involved in coexistence patterns of methanogenic archaeal communities were represented as pairwise links in co-occurrence networks.The network topological properties, which were positively correlated with mean annual temperature, were the most important predictor of CH emissions among all the biotic and abiotic factors. The methanogenic groups involved in commonly co-occurring links among the 39 local networks contributed most to CH emission (53.3%), much higher than the contribution of methanogenic groups with endemic links (36.8%). The potential keystone taxa, belonging to Methanobacterium, Methanocella, Methanothrix, and Methanosarcina, possessed high linkages with the methane generation functional genes mcrA, fwdB, mtbA, and mtbC. Moreover, the commonly coexisting taxa showed a very different assembly pattern, with ~ 30% determinism and ~ 70% stochasticity. In contrast, a higher proportion of stochasticity (93~99%) characterized the assembly of endemically coexisting taxa.These results suggest that the coexistence patterns of microbes are closely tied to their functional significance, and the potential importance of common coexistence further imply that complex networks of interactions may contribute more than species diversity to soil functions. Video abstract.
Impact of urbanization on soil microbial diversity and composition in the megacity of Shanghai
DOI:10.1002/ldr.4145 URL [本文引用: 1]
Critical transition of soil bacterial diversity and composition triggered by nitrogen enrichment
Experimental evidence for a phylogenetic Janzen- Connell effect in a subtropical forest
DOI:10.1111/j.1461-0248.2011.01715.x URL [本文引用: 1]
Plant functional traits—Concepts, applications and future directions
植物功能性状研究进展
Scaling laws predict global microbial diversity
A census-based estimate of Earth’s bacterial and archaeal diversity
DOI:10.1371/journal.pbio.3000106 URL [本文引用: 1]
Increasing aridity reduces soil microbial diversity and abundance in global drylands
Significant impacts of increasing aridity on the arid soil microbiome
Global biogeography of microbial nitrogen-cycling traits in soil
Decreases in average bacterial community rRNA operon copy number during succession
DOI:10.1038/ismej.2015.191 URL [本文引用: 1]
Patterns and processes of microbial community assembly
DOI:10.1128/MMBR.00051-12
PMID:24006468
[本文引用: 2]
Recent research has expanded our understanding of microbial community assembly. However, the field of community ecology is inaccessible to many microbial ecologists because of inconsistent and often confusing terminology as well as unnecessarily polarizing debates. Thus, we review recent literature on microbial community assembly, using the framework of Vellend (Q. Rev. Biol. 85:183-206, 2010) in an effort to synthesize and unify these contributions. We begin by discussing patterns in microbial biogeography and then describe four basic processes (diversification, dispersal, selection, and drift) that contribute to community assembly. We also discuss different combinations of these processes and where and when they may be most important for shaping microbial communities. The spatial and temporal scales of microbial community assembly are also discussed in relation to assembly processes. Throughout this review paper, we highlight differences between microbes and macroorganisms and generate hypotheses describing how these differences may be important for community assembly. We end by discussing the implications of microbial assembly processes for ecosystem function and biodiversity.
FUNGuild: An open annotation tool for parsing fungal community datasets by ecological guild
DOI:10.1016/j.funeco.2015.06.006 URL [本文引用: 2]
Consistent changes in the taxonomic structure and functional attributes of bacterial communities during primary succession
DOI:10.1038/s41396-018-0076-2 URL [本文引用: 1]
Dispersal of arbuscular mycorrhizal fungi: Evidence and insights for ecological studies
DOI:10.1007/s00248-020-01582-x URL [本文引用: 1]
Dimensions of biodiversity in the earth mycobiome
DOI:10.1038/nrmicro.2016.59
PMID:27296482
[本文引用: 1]
Fungi represent a large proportion of the genetic diversity on Earth and fungal activity influences the structure of plant and animal communities, as well as rates of ecosystem processes. Large-scale DNA-sequencing datasets are beginning to reveal the dimensions of fungal biodiversity, which seem to be fundamentally different to bacteria, plants and animals. In this Review, we describe the patterns of fungal biodiversity that have been revealed by molecular-based studies. Furthermore, we consider the evidence that supports the roles of different candidate drivers of fungal diversity at a range of spatial scales, as well as the role of dispersal limitation in maintaining regional endemism and influencing local community assembly. Finally, we discuss the ecological mechanisms that are likely to be responsible for the high heterogeneity that is observed in fungal communities at local scales.
Measuring ectomycorrhizal fungal dispersal: Macroecological patterns driven by microscopic propagules
DOI:10.1111/j.1365-294X.2012.05666.x
PMID:22703050
[本文引用: 1]
Dispersal plays a prominent role in most conceptual models of community assembly. However, direct measurement of dispersal across a whole community is difficult at ecologically relevant spatial scales. For cryptic organisms, such as fungi and bacteria, the scale and importance of dispersal limitation has become a major point of debate. We use an experimental island biogeographic approach to measure the effects of dispersal limitation on the ecological dynamics of an important group of plant symbionts, ectomycorrhizal fungi. We manipulated the isolation of uncolonized host seedlings across a natural landscape and used a range of molecular techniques to measure the dispersal rates of ectomycorrhizal propagules and host colonization. Some species were prolific dispersers, producing annual spore loads on the order of trillions of spores per km(2). However, fungal propagules reaching host seedlings decreased rapidly with increasing distance from potential spore sources, causing a concomitant reduction in ectomycorrhizal species richness, host colonization and host biomass. There were also strong differences in dispersal ability across species, which correlated well with the predictable composition of ectomycorrhizal communities associated with establishing pine forest. The use of molecular tools to measure whole community dispersal provides a direct confirmation for a key mechanism underlying island biogeography theory and has the potential to make microbial systems a model for understanding the role of dispersal in ecological theory.© 2012 Blackwell Publishing Ltd.
Microbial community assembly in marine sediments
DOI:10.3354/ame01826 URL [本文引用: 1]
Toxicity drives facilitation between 4 bacterial species
FungalTraits: A user-friendly traits database of fungi and fungus-like stramenopiles
Host-associated bacterial community succession during amphibian development
DOI:10.1111/mec.14507
PMID:29411448
[本文引用: 1]
Amphibians undergo significant developmental changes during their life cycle, as they typically move from a primarily aquatic environment to a more terrestrial one. Amphibian skin is a mucosal tissue that assembles communities of symbiotic microbiota. However, it is currently not well understood as to where amphibians acquire their skin symbionts, and whether the sources of microbial symbionts change throughout development. In this study, we utilized data collected from four wild boreal toad populations (Anaxyrus boreas); specifically, we sampled the skin bacterial communities during toad development, including eggs, tadpoles, subadults and adults as well as environmental sources of bacteria (water, aquatic sediment and soil). Using 16S rRNA marker gene profiling coupled with SourceTracker, we show that while primary environmental sources remained constant throughout the life cycle, secondary sources of boreal toad symbionts significantly changed with development. We found that toad skin communities changed predictably across development and that two developmental disturbance events (egg hatching and metamorphosis) dictated major changes. Toad skin communities assembled to alternative stable states following each of these developmental disturbances. Using the predicted average rRNA operon copy number of the communities at each life stage, we showed how the skin bacterial communities undergo a successional pattern whereby "fast-growing" (copiotroph) generalist bacteria dominate first before "slow-growing" (oligotroph) specialized bacteria take over. Our study highlights how host-associated bacterial community assembly is tightly coupled to host development and that host-associated communities demonstrate successional patterns akin to those observed in free-living bacteria as well as macrofaunal communities.© 2018 John Wiley & Sons Ltd.
Global phage diversity
Ten new mycobacteriophage genomes presented by show that most phage diversity remains uncharacterized. Extrapolation suggests that less than 0.0002% of the global phage metagenome has been sampled. The new genomes also contain a number of potential virulence factors that may be important in pathogenesis.
Can we predict keystones?
DOI:10.1038/s41579-018-0132-y PMID:30542201 [本文引用: 1]
The community ecology of pathogens: Coinfection, coexistence and community composition
DOI:10.1111/ele.12418
PMID:25728488
[本文引用: 1]
Disease and community ecology share conceptual and theoretical lineages, and there has been a resurgence of interest in strengthening links between these fields. Building on recent syntheses focused on the effects of host community composition on single pathogen systems, we examine pathogen (microparasite) communities using a stochastic metacommunity model as a starting point to bridge community and disease ecology perspectives. Such models incorporate the effects of core community processes, such as ecological drift, selection and dispersal, but have not been extended to incorporate host-pathogen interactions, such as immunosuppression or synergistic mortality, that are central to disease ecology. We use a two-pathogen susceptible-infected (SI) model to fill these gaps in the metacommunity approach; however, SI models can be intractable for examining species-diverse, spatially structured systems. By placing disease into a framework developed for community ecology, our synthesis highlights areas ripe for progress, including a theoretical framework that incorporates host dynamics, spatial structuring and evolutionary processes, as well as the data needed to test the predictions of such a model. Our synthesis points the way for this framework and demonstrates that a deeper understanding of pathogen community dynamics will emerge from approaches working at the interface of disease and community ecology. © 2015 John Wiley & Sons Ltd/CNRS.
Effect of warming and drought on grassland microbial communities
DOI:10.1038/ismej.2011.32 URL [本文引用: 1]
Contrasting elevational diversity patterns between eukaryotic soil microbes and plants
DOI:10.1890/14-0310.1 URL [本文引用: 1]
FungalRoot: Global online database of plant mycorrhizal associations
Estimating and mapping ecological processes influencing microbial community assembly
DOI:10.3389/fmicb.2015.00370
PMID:25983725
[本文引用: 2]
Ecological community assembly is governed by a combination of (i) selection resulting from among-taxa differences in performance; (ii) dispersal resulting from organismal movement; and (iii) ecological drift resulting from stochastic changes in population sizes. The relative importance and nature of these processes can vary across environments. Selection can be homogeneous or variable, and while dispersal is a rate, we conceptualize extreme dispersal rates as two categories; dispersal limitation results from limited exchange of organisms among communities, and homogenizing dispersal results from high levels of organism exchange. To estimate the influence and spatial variation of each process we extend a recently developed statistical framework, use a simulation model to evaluate the accuracy of the extended framework, and use the framework to examine subsurface microbial communities over two geologic formations. For each subsurface community we estimate the degree to which it is influenced by homogeneous selection, variable selection, dispersal limitation, and homogenizing dispersal. Our analyses revealed that the relative influences of these ecological processes vary substantially across communities even within a geologic formation. We further identify environmental and spatial features associated with each ecological process, which allowed mapping of spatial variation in ecological-process-influences. The resulting maps provide a new lens through which ecological systems can be understood; in the subsurface system investigated here they revealed that the influence of variable selection was associated with the rate at which redox conditions change with subsurface depth.
Global diversity and geography of soil fungi
DOI:10.1126/science.1256688 URL [本文引用: 5]
General latitudinal gradient of biodiversity is reversed in ectomycorrhizal fungi
DOI:10.1111/j.1469-8137.2009.03134.x PMID:20088976 [本文引用: 1]
Current insights into phage biodiversity and biogeography
DOI:10.1016/j.mib.2009.08.008
PMID:19811946
[本文引用: 1]
Phages exert tremendous ecological and evolutionary forces directly on their bacterial hosts. Phage induced cell lysis also indirectly contributes to organic and inorganic nutrient recycling. Phage abundance, diversity, and distribution are therefore important parameters in ecosystem function. The assumption that phage consortia are ubiquitous and homogenous across habitats (everything is everywhere) is currently being re-evaluated. New studies on phage biogeography have found that some phages are globally distributed while others are unique and perhaps endemic to specific environments. Furthermore, advances in technology have allowed scientists to conduct experiments aimed at analyzing phage consortia over temporal scales, and surprisingly have found reoccurring patterns. This review discusses currents in the field of phage ecology with particular focus on efforts to characterize phage diversity and biogeography across various spatial and temporal scales.
Soil organic matter availability and climate drive latitudinal patterns in bacterial diversity from tropical to cold temperate forests
DOI:10.1111/1365-2435.12952 URL [本文引用: 1]
Fungal traits that drive ecosystem dynamics on land
DOI:10.1128/MMBR.00001-15
PMID:25971588
[本文引用: 1]
Fungi contribute extensively to a wide range of ecosystem processes, including decomposition of organic carbon, deposition of recalcitrant carbon, and transformations of nitrogen and phosphorus. In this review, we discuss the current knowledge about physiological and morphological traits of fungi that directly influence these processes, and we describe the functional genes that encode these traits. In addition, we synthesize information from 157 whole fungal genomes in order to determine relationships among selected functional genes within fungal taxa. Ecosystem-related traits varied most at relatively coarse taxonomic levels. For example, we found that the maximum amount of variance for traits associated with carbon mineralization, nitrogen and phosphorus cycling, and stress tolerance could be explained at the levels of order to phylum. Moreover, suites of traits tended to co-occur within taxa. Specifically, the genetic capacities for traits that improve stress tolerance-β-glucan synthesis, trehalose production, and cold-induced RNA helicases-were positively related to one another, and they were more evident in yeasts. Traits that regulate the decomposition of complex organic matter-lignin peroxidases, cellobiohydrolases, and crystalline cellulases-were also positively related, but they were more strongly associated with free-living filamentous fungi. Altogether, these relationships provide evidence for two functional groups: stress tolerators, which may contribute to soil carbon accumulation via the production of recalcitrant compounds; and decomposers, which may reduce soil carbon stocks. It is possible that ecosystem functions, such as soil carbon storage, may be mediated by shifts in the fungal community between stress tolerators and decomposers in response to environmental changes, such as drought and warming. Copyright © 2015, American Society for Microbiology. All Rights Reserved.
Soil pH mediates the balance between stochastic and deterministic assembly of bacteria
DOI:10.1038/s41396-018-0082-4 URL [本文引用: 1]
Community assembly and coexistence in communities of arbuscular mycorrhizal fungi
DOI:10.1038/ismej.2016.46 URL [本文引用: 1]
Environment and host as large-scale controls of ectomycorrhizal fungi
DOI:10.1038/s41586-018-0189-9 URL [本文引用: 2]
Temporal tracking of quantum-dot apatite across in vitro mycorrhizal networks shows how host demand can influence fungal nutrient transfer strategies
DOI:10.1038/s41396-020-00786-w URL [本文引用: 1]
Nutrient dependent cross-kingdom interactions: Fungi and bacteria from an oligotrophic desert oasis
DOI:10.3389/fmicb.2018.01755
PMID:30131780
[本文引用: 1]
Microbial interactions play a key role in ecosystem functioning, with nutrient availability as an important determinant. Although phylogenetically distant bacteria and fungi commonly co-occur in nature, information on their cross-kingdom interactions under unstable, extreme environments remains poor. Hence, the aims of this work were to evaluate potential in vitro interactions among fungi and bacteria isolated from a phosphorous oligotrophic aquatic system in the Cuatro Cienegas Basin, Mexico, and to test the nutrients-based shifts. We assessed growth changes in bacteria (Aeromonas and Vibrio) and fungi (Coprinellus micaceus, Cladosporium sp., and Aspergillus niger) on co-cultures in relation to monocultures under diverse nutrient scenarios on Petri dishes. Interactions were explored using a network analysis, and a metabolome profiling for specific taxa. We identified nutrient-dependent patterns, as beneficial interactions dominated in low-nutrients media and antagonistic interactions dominated in rich media. This suggests that cross-kingdom synergistic interactions might favor microbial colonization and growth under low nutrient conditions, representing an adaptive trait to oligotrophic environments. Moreover, our findings agree with the stress-gradient hypothesis, since microbial interactions shifted from competition to cooperation as environmental stress (expressed as low nutrients) increased. At a functional level consistent differences were detected in the production of secondary metabolites, agreeing with plate bioassays. Our results based on culture experiments, provides evidence to understand the complexity of microbial dynamics and survival in phosphorous-depleted environments.
Conceptual synthesis in community ecology
DOI:10.1086/652373 URL [本文引用: 3]
Assessing the relative importance of neutral stochasticity in ecological communities
DOI:10.1111/oik.01493 URL [本文引用: 1]
A meta-analysis of global fungal distribution reveals climate-driven patterns
DOI:10.1038/s41467-019-13164-8
PMID:31723140
[本文引用: 1]
The evolutionary and environmental factors that shape fungal biogeography are incompletely understood. Here, we assemble a large dataset consisting of previously generated mycobiome data linked to specific geographical locations across the world. We use this dataset to describe the distribution of fungal taxa and to look for correlations with different environmental factors such as climate, soil and vegetation variables. Our meta-study identifies climate as an important driver of different aspects of fungal biogeography, including the global distribution of common fungi as well as the composition and diversity of fungal communities. In our analysis, fungal diversity is concentrated at high latitudes, in contrast with the opposite pattern previously shown for plants and other organisms. Mycorrhizal fungi appear to have narrower climatic tolerances than pathogenic fungi. We speculate that climate change could affect ecosystem functioning because of the narrow climatic tolerances of key fungal taxa.
Co-occurrence of planktonic bacteria and archaea affects their biogeographic patterns in China’s coastal wetlands
DOI:10.1186/s40793-021-00388-9 URL [本文引用: 1]
Archaea is more important than bacteria in driving soil stoichiometry in phosphorus deficient habitats
DOI:10.1016/j.scitotenv.2022.154417 URL [本文引用: 1]
Higher precipitation strengthens the microbial interactions in semi- arid grassland soils
DOI:10.1111/geb.12718 URL [本文引用: 1]
The Anthropocene is functionally and stratigraphically distinct from the Holocene
Trait dimensions in bacteria and archaea compared to vascular plants
DOI:10.1111/ele.13742 URL [本文引用: 1]
Trading on the arbuscular mycorrhiza market: From arbuscules to common mycorrhizal networks
DOI:10.1111/nph.15775
PMID:30843207
[本文引用: 1]
Arbuscular mycorrhiza (AM) symbiosis occurs between obligate biotrophic fungi of the phylum Glomeromycota and most land plants. The exchange of nutrients between host plants and AM fungi (AMF) is presumed to be the main benefit for the two symbiotic partners. In this review article, we outline the current concepts of nutrient exchanges within this symbiosis (mechanisms and regulation). First, we focus on phosphorus and nitrogen transfer from the fungal partner to the host plant, and on the reciprocal transfer of carbon compounds, with a highlight on a possible interplay between nitrogen and phosphorus nutrition during AM symbiosis. We further discuss potential mechanisms of regulation of these nutrient exchanges linked to membrane dynamics. The review finally addresses the common mycorrhizal networks formed AMF, which interconnect plants from similar and/or different species. Finally the best way to integrate this knowledge and the ensuing potential benefits of AM into sustainable agriculture is discussed.© 2019 The Authors. New Phytologist © 2019 New Phytologist Trust.
Reduction of microbial diversity in grassland soil is driven by long-term climate warming
DOI:10.1038/s41564-022-01147-3 URL [本文引用: 1]
Genome-resolved metagenomics reveals role of iron metabolism in drought-induced rhizosphere microbiome dynamics
DOI:10.1038/s41467-021-23553-7
PMID:34050180
[本文引用: 1]
Recent studies have demonstrated that drought leads to dramatic, highly conserved shifts in the root microbiome. At present, the molecular mechanisms underlying these responses remain largely uncharacterized. Here we employ genome-resolved metagenomics and comparative genomics to demonstrate that carbohydrate and secondary metabolite transport functionalities are overrepresented within drought-enriched taxa. These data also reveal that bacterial iron transport and metabolism functionality is highly correlated with drought enrichment. Using time-series root RNA-Seq data, we demonstrate that iron homeostasis within the root is impacted by drought stress, and that loss of a plant phytosiderophore iron transporter impacts microbial community composition, leading to significant increases in the drought-enriched lineage, Actinobacteria. Finally, we show that exogenous application of iron disrupts the drought-induced enrichment of Actinobacteria, as well as their improvement in host phenotype during drought stress. Collectively, our findings implicate iron metabolism in the root microbiome's response to drought and may inform efforts to improve plant drought tolerance to increase food security.
Drought delays development of the Sorghum root microbiome and enriches for monoderm bacteria
Phylogenetic imprint of woody plants on the soil mycobiome in natural mountain forests of Eastern China
DOI:10.1038/s41396-018-0303-x URL [本文引用: 1]
Emerging patterns of microbial functional traits
DOI:10.1016/j.tim.2021.04.004 URL [本文引用: 1]
Fungal functional ecology: Bringing a trait-based approach to plant-associated fungi
DOI:10.1111/brv.12570
[本文引用: 1]
Fungi play many essential roles in ecosystems. They facilitate plant access to nutrients and water, serve as decay agents that cycle carbon and nutrients through the soil, water and atmosphere, and are major regulators of macro-organismal populations. Although technological advances are improving the detection and identification of fungi, there still exist key gaps in our ecological knowledge of this kingdom, especially related to function. Trait-based approaches have been instrumental in strengthening our understanding of plant functional ecology and, as such, provide excellent models for deepening our understanding of fungal functional ecology in ways that complement insights gained from traditional and -omics-based techniques. In this review, we synthesize current knowledge of fungal functional ecology, taxonomy and systematics and introduce a novel database of fungal functional traits (Fun(Fun)). Fun(Fun) is built to interface with other databases to explore and predict how fungal functional diversity varies by taxonomy, guild, and other evolutionary or ecological grouping variables. To highlight how a quantitative trait-based approach can provide new insights, we describe multiple targeted examples and end by suggesting next steps in the rapidly growing field of fungal functional ecology.
Soil available phosphorus content drives the spatial distribution of archaeal communities along elevation in acidic terrace paddy soils
DOI:10.1016/j.scitotenv.2018.12.144
[本文引用: 1]
Archaea play crucial roles in geochemical cycles and influence the emission of greenhouse gases in acidic soils. However, little is known about the distribution pattern of total archaeal diversity and community composition with increasing elevation, especially in acidic agricultural ecosystems. Terraces, characterized by vertical climate changes and unique hydrological properties, are "natural experiments" to explore the spatial distribution of microorganisms along elevation in paddy soils. Here we investigated the diversify and structure of soil archaeal communities in nine increasingly elevated acidic paddy soils of the Yunhe terrace, China. Archaeal communities were dominated by Methanomicrobia of Euryarchaeota (38.5%), Group 1,1a-associated cluster (SAGSCG-1) of Thaumarchaeota (22.0%) and Subgroup-6 (previously described as crenarchaeotal group 1.3b) of Bathyarchaeota (17.8%). The archaeal phylotype richness decreased with increasing elevation. Both the species richness and phylogenetic diversity of the archaeal communities were significantly negatively correlated with soil available phosphorus (AP) content according to linear regression analyses. The archaeal communities differed greatly between soils of increasing elevation, and were roughly clustered into three groups, mostly in relation to AP contents. A variation partitioning analysis further confirmed that edaphic factors including the content of AP (17.1%), nitrate (7.83%), soil organic carbon (4.69%), dissolved organic carbon (4.22%) and soil pH (4.07%) shaped the archaeal community. The variation of soil properties were probably induced by elevation. The co-occurrence network indicated a modular structure of the archaeal community. Overall, our results emphasized that soil AP content. was the best predictor of archaeal diversify and community structure, and the impacts of elevation on soil archaeal communities were no diminished by long hum rice cultivation, although minor compared with the effects of soil properties. (C) 2018 Elsevier B.V.
Environmental changes affect the assembly of soil bacterial community primarily by mediating stochastic processes
DOI:10.1111/gcb.13080
PMID:26340501
[本文引用: 1]
Both 'species fitness difference'-based deterministic processes, such as competitive exclusion and environmental filtering, and 'species fitness difference'-independent stochastic processes, such as birth/death and dispersal/colonization, can influence the assembly of soil microbial communities. However, how both types of processes are mediated by anthropogenic environmental changes has rarely been explored. Here we report a novel and general pattern that almost all anthropogenic environmental changes that took place in a grassland ecosystem affected soil bacterial community assembly primarily through promoting or restraining stochastic processes. We performed four experiments mimicking 16 types of environmental changes and separated the compositional variation of soil bacterial communities caused by each environmental change into deterministic and stochastic components, with a recently developed method. Briefly, because the difference between control and treatment communities is primarily caused by deterministic processes, the deterministic change was quantified as (mean compositional variation between treatment and control) - (mean compositional variation within control). The difference among replicate treatment communities is primarily caused by stochastic processes, so the stochastic change was estimated as (mean compositional variation within treatment) - (mean compositional variation within control). The absolute of the stochastic change was greater than that of the deterministic change across almost all environmental changes, which was robust for both taxonomic and functional-based criterion. Although the deterministic change may become more important as environmental changes last longer, our findings showed that changes usually occurred through mediating stochastic processes over 5 years, challenging the traditional determinism-dominated view. © 2015 John Wiley & Sons Ltd.
Biogeography, assembly patterns, driving factors, and interactions of archaeal community in mangrove sediments
DOI:10.1128/mSystems.01381-20 URL [本文引用: 1]
Assembly processes lead to divergent soil fungal communities within and among 12 forest ecosystems along a latitudinal gradient
DOI:10.1111/nph.17457
PMID:33982802
[本文引用: 1]
Latitudinal gradients provide opportunities to better understand soil fungal community assembly and its relationship with vegetation, climate, soil and ecosystem function. Understanding the mechanisms underlying community assembly is essential for predicting compositional responses to changing environments. We quantified the relative importance of stochastic and deterministic processes in structuring soil fungal communities using patterns of community dissimilarity observed within and between twelve natural forests and related these to environmental variation within and among sites. The results revealed that whole fungal communities and communities of arbuscular and ectomycorrhizal fungi consistently exhibited divergent patterns but with less divergence for ectomycorrhizal fungi at most sites. Within those forests, no clear relationships were observed between the degree of divergence within fungal and plant communities. When comparing communities at larger spatial scales, among the twelve forests, we observed distinct separation in all three fungal groups among tropical, subtropical and temperate climatic zones. Soil fungal β-diversity patterns between forests were also greater when comparing forests exhibiting high environmental heterogeneity. Taken together, although large-scale community turnover could be attributed to specific environmental drivers, the differences among fungal communities in soils within forests was high even at local scales.This article is protected by copyright. All rights reserved.
Temperature mediates continental-scale diversity of microbes in forest soils
DOI:10.1038/ncomms12083
PMID:27377774
[本文引用: 1]
Climate warming is increasingly leading to marked changes in plant and animal biodiversity, but it remains unclear how temperatures affect microbial biodiversity, particularly in terrestrial soils. Here we show that, in accordance with metabolic theory of ecology, taxonomic and phylogenetic diversity of soil bacteria, fungi and nitrogen fixers are all better predicted by variation in environmental temperature than pH. However, the rates of diversity turnover across the global temperature gradients are substantially lower than those recorded for trees and animals, suggesting that the diversity of plant, animal and soil microbial communities show differential responses to climate change. To the best of our knowledge, this is the first study demonstrating that the diversity of different microbial groups has significantly lower rates of turnover across temperature gradients than other major taxa, which has important implications for assessing the effects of human-caused changes in climate, land use and other factors.
Stochasticity, succession, and environmental perturbations in a fluidic ecosystem
Stochastic community assembly: Does it matter in microbial ecology?
Meta-analysis of the impacts of global change factors on soil microbial diversity and functionality
DOI:10.1038/s41467-020-16881-7
PMID:32555185
[本文引用: 2]
Biodiversity on the Earth is changing at an unprecedented rate due to a variety of global change factors (GCFs). However, the effects of GCFs on microbial diversity is unclear despite that soil microorganisms play a critical role in biogeochemical cycling. Here, we synthesize 1235 GCF observations worldwide and show that microbial rare species are more sensitive to GCFs than common species, while GCFs do not always lead to a reduction in microbial diversity. GCFs-induced shifts in microbial alpha diversity can be predominately explained by the changed soil pH. In addition, GCF impacts on soil functionality are explained by microbial community structure and biomass rather than the alpha diversity. Altogether, our findings of GCF impacts on microbial diversity are fundamentally different from previous knowledge for well-studied plant and animal communities, and are crucial to policy-making for the conservation of microbial diversity hotspots under global changes.

{kind=link}
{kind=link}
{kind=link}
{kind=link}