
|
|
||
|
巴尔通体在滇西南蝙蝠中高度流行并具有丰富的遗传变异特征
生物多样性
2021, 29 (9):
1245-1255.
DOI: 10.17520/biods.2021028
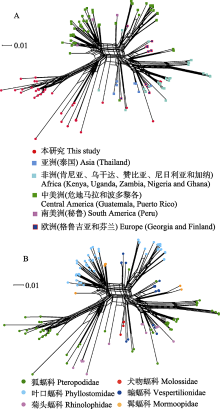
蝙蝠是很多病原微生物的自然宿主, 全球多项研究表明蝙蝠是巴尔通体(Bartonella species)的主要宿主。为了解滇西南地区蝙蝠中巴尔通体的流行特征, 我们于2015-2017年间在云南省4个地区应用网捕法捕获蝙蝠3种305只。经种类鉴定后采集肝脾组织, 提取核酸, 通过TaqMan实时荧光定量PCR方法检测巴尔通体的tmRNA基因ssrA, 并进行测序鉴定和系统发育分析。结果发现172只蝙蝠检出该基因, 总感染率为56.4%; 其中临沧、西双版纳、保山和瑞丽4个采样点的蝙蝠感染率分别为50.0% (22/44)、61.7% (29/47)、62.1% (18/29)和55.7% (103/185)。中菊头蝠(Rhinolophus affinis)、小菊头蝠(R. blythi)和棕果蝠(Rousettus leschenaultii)的感染率分别为50.0% (22/44)、62.1% (18/29)和56.9% (132/232), 差异没有统计学意义(χ2 = 1.135, P = 0.567), 表明巴尔通体在云南当地的蝙蝠种群中高度流行。定量PCR扩增产物2次扩增后测序获得37个巴尔通体ssrA序列, 属于10个系统发育分支, 其中1个为伊丽莎白巴尔通体(B. elizabethae)、特利波契巴尔通体(B. tribocorum)和克拉斯诺夫巴尔通体(B. krasnovii)的近缘种。其余序列与已知巴尔通体距离较远, 与亚洲、欧洲和美洲等其他地域来源于蝙蝠的巴尔通体近缘。遗传多样性分析显示, ssrA基因的核苷酸多样性指数(π)为0.11381 ± 0.00928, 基因型多样性指数(Hd)为0.985 ± 0.010, 形成29个基因型(单倍型), 说明云南蝙蝠巴尔通体具有丰富的遗传多样性。通过对本研究标本与全球相关序列的系统发育网络重建, 分析全球蝙蝠巴尔通体的地理和宿主分布特征, 可以看出巴尔通体与蝙蝠之间存在显著的宿主特异性关联。因此可初步确定蝙蝠-巴尔通体具有协同进化特征, 同时受到地理隔离的影响。 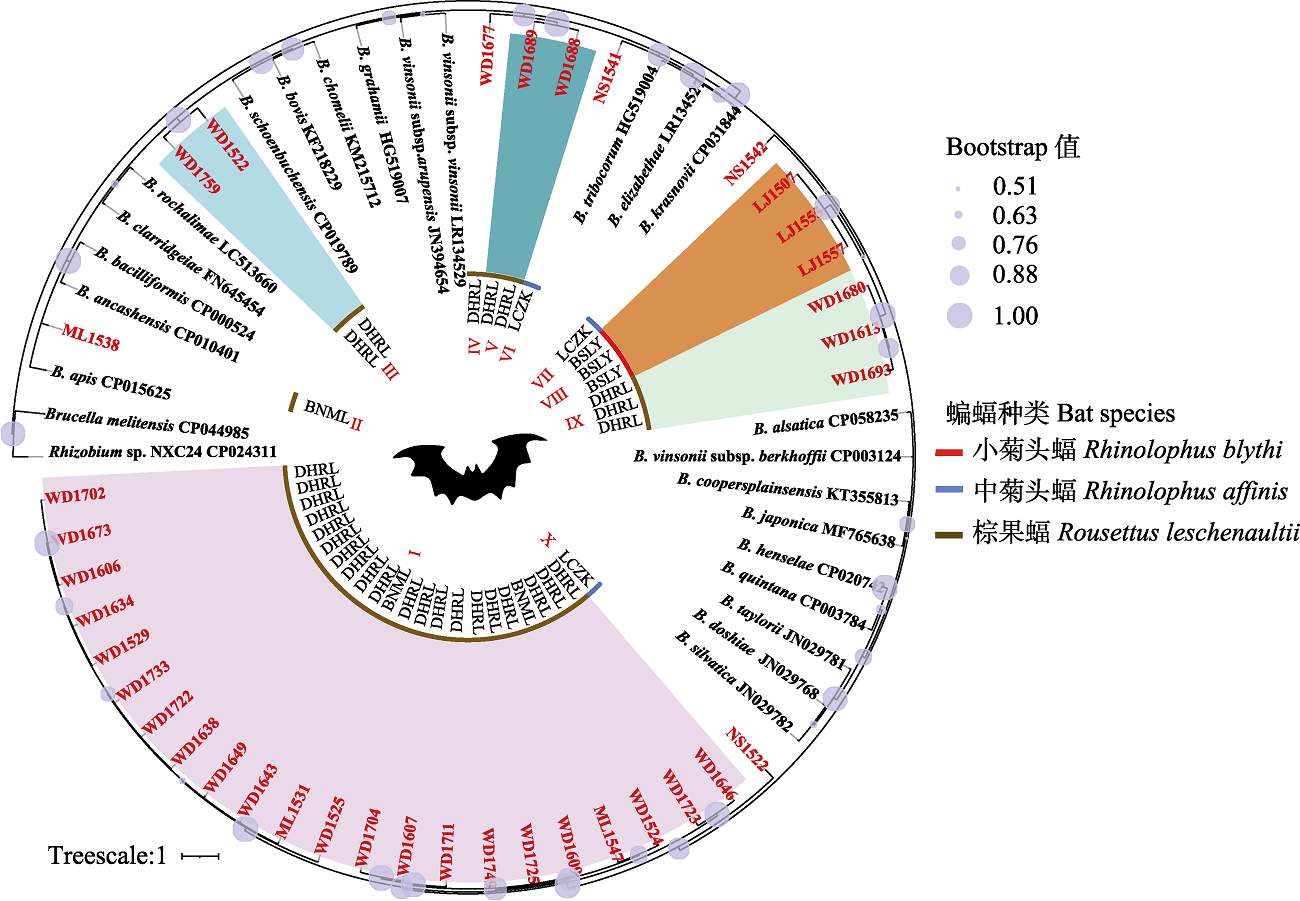 View image in article
图1
基于蝙蝠标本及巴尔通体参考菌株的ssrA序列构建的贝叶斯系统发育树。蝙蝠种类在系统树中内圈, 以红、蓝和棕色线段表示; 本研究中蝙蝠序列名称用红色字体标识, 同附录3中的标本名称。图中心BSLY、LCZK、BNML和DHRL分别代表保山市隆阳区潞江镇、临沧市镇康县南伞乡、西双版纳州勐腊县瑶区乡和德宏州瑞丽市畹町镇; 图中心Ⅰ-X代表本研究中蝙蝠ssrA序列所在的进化分支; Brucella melitensis CP044985和Rhizobuim sp. NXC24 CP024311为外群。
正文中引用本图/表的段落
在ssrA序列的系统发育树中(图1), 本研究中的37个蝙蝠巴尔通体序列分布到10个分支中, 用I-X表示。其中, 分支I、III、V、VIII和IX拓扑位置稳定, 分支节点的后验概率是100%; IV、VI和X的拓扑位置亦属可信, 分支节点的后验概率大于90%; 分支II和VII在树中没有稳定结构关系, 后验概率小于60%。在本研究序列中, 处于分支VI的NS1541与伊丽莎白巴尔通体(B. elizabethae)、特利波契巴尔通体和克拉斯诺夫巴尔通体(B. krasnovii)亲缘关系较近, 聚在一起, 由于序列相似值较低(附录3), 不能确认是哪种巴尔通体; 剩余其他分支的序列均与已知巴尔通体距离较远, 序列相似值较低, 无法确定巴尔通体种类。从系统发育树中还可以看出, 分支I、III、V、VIII和IX分别是来自相同或不同地区的同种蝙蝠宿主的巴尔通体序列, 呈现出聚集性, 如分支I包括来自西双版纳州和德宏州的棕果蝠巴尔通体序列, 分支VIII包括来自保山市的小菊头蝠巴尔通体。
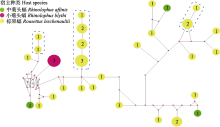
37条包含278 bp的ssrA序列代表37株蝙蝠巴尔通体, 共检测到106个多态位点、27个单变异位点、79个简约信息位点, 生成29个基因型, 核苷酸多样性指数(π)为0.11381 ± 0.00928, 基因型多样性指数(Hd)为0.985 ± 0.010, 平均核苷酸差异数(k)为27.883, 中性检验Tajima's D值为?0.73641 (P > 0.10)。由29个基因型构建的MJ网络图呈现出较为复杂的拓扑结构(图2), 3株小菊头蝠形成一个基因型; 3株中菊头蝠形成3个基因型, 散落在整个网络中, 距离较远, 这与二叉树(图1)中的结构一致; 31株棕果蝠形成了25个基因型, 只有3个明确的系统发生关系(图中用蓝色虚线框标识), 其余基因型均被mv (median vectors)即假想的共同祖先序列分隔开来。
本文的其它图/表
|
{kind=link}