



植物不同器官的外部和内部栖居着数量庞大且种类繁多的微生物, 包括细菌、古菌、真菌、原生动物和病毒等, 这些微生物及其遗传信息和代谢产物等共同组成了植物微生物组(plant microbiomes) (Müller et al, 2016; Trivedi et al, 2020)。据估计植物微生物基因组总和远远超过其宿主基因组, 被称为植物的“第二基因组” (Berendsen et al, 2012)。微生物与其宿主植物作为“共生功能体” (holobiont), 在植物养分吸收、生长发育、病害抵御和环境胁迫适应性等方面发挥了重要作用(Sánchez-Cañizares et al, 2017)。同时, 植物微生物组中也存在大量致病性微生物, 包括病毒、细菌、真菌和卵菌等, 在病原体大量繁殖的条件下可引起植物病害和生态系统失衡(Mendes et al, 2013)。在当今耕地退化、环境污染和气候变化等多重挑战下, 充分认识并挖掘植物微生物组资源对地上地下生物多样性保护、生态系统管理和农业可持续发展具有重要意义。
得益于多组学技术的发展应用, 近10多年来植物微生物组研究取得了一系列进展。基于模式植物拟南芥(Arabidopsis thaliana)、主要农作物如玉米(Zea mays)、水稻(Oryza sativa)、小麦(Triticum aestivum)等以及木本植物如柑橘(Citrus)、杨树(Populus)等的研究极大促进了我们对植物微生物组多样性、组成和功能特征的认识。如对植物微生物的认识从传统的细菌、真菌扩展到更多生物类群以及生物间的相互作用, 揭示了原生生物、噬菌体等通过调节微生物群落间的互作关系进而影响植物健康和生长发育的功能作用等(Morella et al, 2018; Guo et al, 2022)。并从植物-微生物互作角度揭示了微生物促进植物养分吸收和生长发育、提高植物对病原菌及环境胁迫的抵抗能力的作用机制(Carrión et al, 2019; Zhang et al, 2019; de Vries et al, 2020)。此外, 大量研究探讨了生物和非生物因素对植物微生物群落构建过程的影响, 并开展了植物微生物合成群落的开发应用探索(Xiong et al, 2021b, c; Zhang et al, 2022)。相关进展为利用植物微生物组功能服务于植物健康维持和生产力提高提供了重要基础。因此, 本文对近年来有关植物微生物群落的多样性和组成特征、功能作用及其机制、植物微生物群落的构建过程和驱动因素等相关研究进展进行了总结论述, 同时对目前存在的问题及未来研究需要加强的发展方向进行了展望, 以期引起人们对植物微生物组研究和开发应用的重视。
1 植物微生物群落多样性及组成特征
植物微生物组中以细菌和真菌的数量和种类最多(图1)。据估计, 每克植物(地上部鲜重)中细菌和真菌的丰度分别可达102-107和101-103基因拷贝数, 多样性达101-105和101-102个OTUs (operational taxonomic units, 可操作分类单元) (Leach et al, 2017)。与土壤细菌群落主要以变形菌门、放线菌门、酸杆菌门和浮霉菌门为主略有不同, 植物相关细菌群落主要由变形菌门、放线菌门、拟杆菌门和厚壁菌门等类群组成(Bai et al, 2015; Edwards et al, 2015; Delgado-Baquerizo et al, 2018)。从土壤到植物根际, 再到根内, 细菌群落多样性逐级下降, 群落组成差异增加, 拟杆菌门、酸杆菌门、绿弯菌门和疣微菌门等显著减少, 变形菌门和厚壁菌门在根内显著富集, 其相对丰度能达到根际的两倍以上(Edwards et al, 2015; Niu et al, 2017; Hamonts et al, 2018; Trivedi et al, 2020)。类似地, 变形菌门占植物叶际细菌群落的50%以上, 并以甲基杆菌属(Methylobacterium)、假单胞菌属(Pseudomonas)、鞘氨醇单胞菌属(Sphingomonas)、节杆菌属(Arthrobacter)和泛菌属(Pantoea)为主(Rastogi et al, 2013) (图1)。
图1
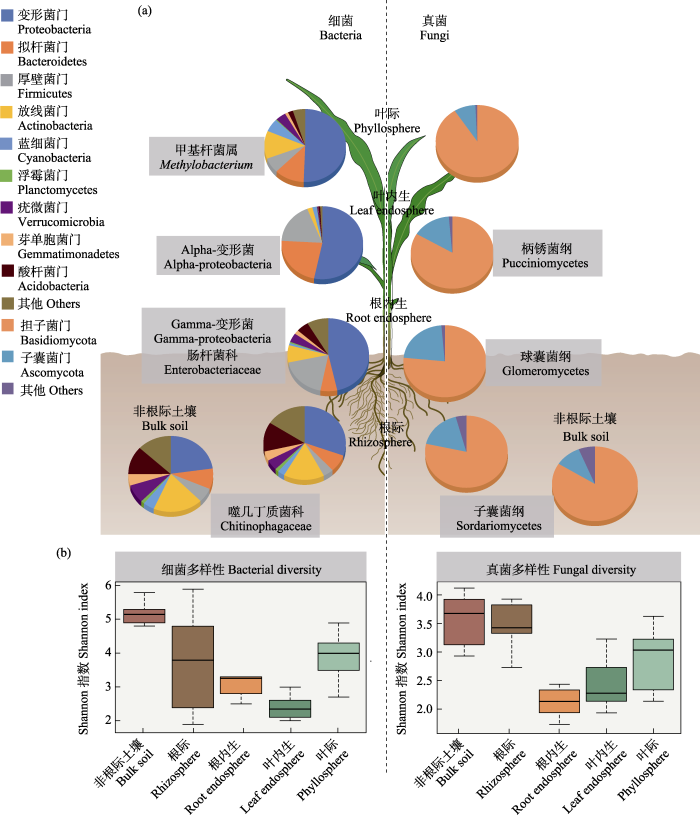
图1
植物不同部位生态位细菌和真菌群落组成(a)和多样性(b)特征。改自Trivedi等(2020)和Xiong等(2021a, b, c)基于玉米、小麦、大麦等植物微生物组研究数据。图a方框中的细菌和真菌类群代表不同部位生态位的指示类群。
Fig. 1
The patterns of plant-associated bacterial and fungal community compositions (a) and diversities (b) across multiple plant compartment niches. Modified from Trivedi et al (2020) and Xiong et al (2021a, b, c) based on data from maize, wheat and barley related studies. The bacterial and fungal taxa in the boxes in
除细菌和真菌外, 植物不同部位还生活着丰富多样的古菌、原生生物和病毒等, 但其丰度和多样性低于细菌和真菌(Vandenkoornhuyse et al, 2015; Toju et al, 2018)。据估计, 每克植物(地上部鲜重) 约含101-102个古菌OTUs, 主要属于奇古菌门、泉古菌门和广古菌门, 且在植物不同部位和不同宿主植物间具有明显差异(Leach et al, 2017; Moissl-Eichinger et al, 2018)。如在植物根系和根际微缺氧环境中分布着大量的产甲烷古菌和氨氧化古菌, 与根际和根区碳氮代谢密切相关(Ke et al, 2014)。多年生植物的叶片内部也被认为是古菌的适宜生境, 如地中海橄榄树(Olea europaea)叶内生古菌的相对丰度高达35.8%, 主要属于奇古菌门和泉古菌门(Müller et al, 2015)。古菌具有适应极端环境条件的特征, 在各种生境下广泛存在, 但目前关于古菌的研究主要是以产甲烷古菌和氨氧化古菌为代表, 而且集中在海洋和土壤环境中(张丽梅和贺纪正, 2012; Zhou et al, 2018)。有关植物地上部和植物内生古菌的特征及其功能作用知之甚少, 特别是古菌的定殖是否会导致植物发生病害仍不清楚, 相关研究亟待开展。
原生生物是指除植物、真菌和动物外的单细胞真核生物, 其可以通过调节食物网中的捕食关系影响植物微生物群落(Geisen et al, 2018; 姚保民等, 2022)。目前有关原生生物的报道主要集中于土壤和根际环境, 如有研究发现柳枝稷(Panicum virgatum)根际原生生物多样性低于非根际土壤, 但属于Colpodida、Flamellidae和Cyrtolophosis的杂食性原生生物以及属于霜霉目的致病性原生生物在根际显著富集(Ceja-Navarro et al, 2021)。叶际原生生物多样性显著低于根际和非根际土壤, 群落组成也与土壤明显不同, 主要属于有孔虫超群、囊泡虫超群和变形虫超群(Sapp et al, 2018; Sun et al, 2021)。此外, 病毒作为地球上数量最多、种类最丰富的生物实体, 除可通过直接感染宿主植物引起病毒病害外, 还可通过裂解优势微生物和携带辅助代谢基因来影响植物微生物组结构和功能(Pratama & van Elsas, 2018)。但以往的研究更多针对引起某一类植物病害的病毒开展, 有关植物微生物组中病毒, 尤其是噬菌体的多样性及其功能还有待深入研究。
2 植物微生物组的生态功能及其作用机制
植物微生物组在促进植物营养吸收、调节植物生长发育、提高植物病害防御能力以及环境胁迫适应性等方面均发挥着重要作用, 而这些功能的实现与植物-微生物之间的互作关系是紧密相关的, 其具体功能和作用机制如下(Box 1)。
2.1 促进植物养分吸收
在营养贫瘠的环境中, 植物微生物群落对植物养分吸收起着重要作用, 如与植物形成紧密共生关系的根瘤菌和菌根真菌。据估计, 豆科植物-根瘤菌共生固氮体系的年固氮量约占全球生物固氮总量的60%-70%, 能满足植物需氮量的90%以上(Richardson et al, 2009)。自然界中80%以上的陆地植物都能与菌根真菌形成共生体系, 菌根真菌通过菌丝增加养分吸收的空间并活化土壤中的难溶性磷, 为植物提供了80%左右的磷元素(van der Heijden et al, 2015, 2017)。有趣的是, 植物共生微生物还可以相互作用以促进植物营养吸收, 如非根际土壤中的根瘤菌可以经内生真菌枫香拟茎点霉(Phomopsis liquidambaris)的菌丝扩散到达花生根际并与根系形成根瘤进行共生固氮(Zhang et al, 2020)。
除了这些能够与植物形成明显共生结构的微生物外, 根表或叶表栖居的微生物对促进宿主养分吸收同样至关重要。在缺氮土壤中, 玉米可以通过分泌黄酮类物质招募草酸杆菌科细菌促进氮的吸收, 或通过气生根分泌黏液招募非共生固氮菌进行生物固氮, 可为玉米生长季提供29%-82%的氮素(van Deynze et al, 2018; Yu et al, 2021)。叶际高碳氮比的环境有利于非共生固氮微生物生存, 在半干旱森林系统中, 从麻疯树(Jatropha curcas)叶际分离到的内生细菌中甲基杆菌属占比69.1%, 其中30.2%能够参与固氮, 因此有学者推测麻疯树对贫瘠土壤的耐受性可能与其叶际固氮能力有关(Lindow & Brandl, 2003; Madhaiyan et al, 2015)。在极度缺磷的高山环境中, 不会形成菌根结构的十字花科植物如高山拟南芥(Arabis alpina)可通过根际富集非菌根类真菌柔膜菌目以增加磷的吸收并促进植物生长(Almario et al, 2017)。此外, 在缺磷条件下拟南芥还可通过免疫反应启动磷酸盐饥饿反应系统控制植物病原真菌炭疽菌(Colletotrichum tofieldiae)在根上定殖从而将大量磷酸盐转移到嫩枝上, 但不引起植株产生疾病(Hiruma et al, 2016)。而在铁饥饿条件下, 拟南芥可通过分泌次级代谢产物如香豆素改变根系微生物组结构以活化环境中的铁或产生活性氧抑制与植物竞争铁营养的假单胞菌大量定殖(Voges et al, 2019; Harbort et al, 2020)。
2.2 调节植物生长发育
微生物可以通过直接产生激素或间接影响植物激素水平调控宿主植物生长发育(Bai et al, 2022)。植物根际或地上部大量存在的非共生固氮菌, 如从中国芒草(Miscanthus sinensis)根内分离得到的内生固氮菌假单胞菌(Pseudomonas sp. Y-5), 其基因组中拥有植物促生相关基因, 能显著提高植物根/茎氮含量和鲜重, 表现出良好的促生能力(Li et al, 2022b)。此外, 一些植物内生菌如红球菌属(Rhodococcus)和黄杆菌属(Flavobacterium)等也可以直接产生生长素(IAA)促进植物根系伸长和植物生长(Belimov et al, 2005)。大量基于拟南芥的研究则发现, 根系细菌如恶臭假单胞菌(Pseudomonas putida UW4)可通过降低植物乙烯水平促进植物生长的同时伴随着强烈的胁迫过敏反应(Ravanbakhsh et al, 2019)。基于合成群落的方法发现贪噬菌属(Variovorax)细菌与拟南芥根系生长密切相关, 其拥有高度保守的IAA降解操纵子, 可以通过调节IAA水平来促进定型根发育(Finkel et al, 2020)。最近研究还发现广泛存在于双子叶植物中的病原真菌核盘菌(Sclerotinia sclerotiorum)可通过修饰小麦抗病和光合作用相关基因的表达, 提高IAA表达水平, 影响宿主植物的生理代谢(Tian et al, 2020)。
2.3 影响植物健康和病害防御
植物微生物组在促进宿主健康和提高病害抵御能力方面也起着重要作用。根际微生物被认为是抵御病原菌入侵的“第一道防线” (Mendes et al, 2018; Gao et al, 2021; Ge et al, 2021; Liu et al, 2021)。植物遭受到病原菌入侵后, 会迅速启动免疫反应, 激活下游与抗病有关基因的表达, 同时植物会向微生物“呼救”以招募有益微生物来抵御病原菌胁迫(Kwak et al, 2018; Ge et al, 2022)。如丁香假单胞菌(Pseudomonas syringae)入侵番茄叶片后, 根系分泌的L-苹果酸会增加, 致使有益菌枯草芽孢杆菌(Bacillus subtilis FB17)在根系富集并形成生物膜(Rudrappa et al, 2008)。不同的尖孢镰刀菌(Fusarium oxysporum)病原小种侵染辣椒(Capsicum annuum)、西瓜(Citrullus lanatus)等植物后会导致根际假单胞菌属、链霉菌属(Streptomyces)和芽孢杆菌属(Bacillus)等具有抗病促生功能的微生物类群显著富集(Gao et al, 2021; Ge et al, 2022)。此外, 根际微生物在抑病性土壤(disease suppressive soil, 指某一土传病害长期暴发后形成的对病原菌具有免疫抗性的土壤)形成中扮演着重要角色(Mendes et al, 2011; 张瑞福和沈其荣, 2012)。如在小麦连作系统中, 全蚀病多次爆发后其症状逐渐减轻, 主要是由于根际荧光假单胞菌(Pseudomonas fluorescens)的定殖并产生抗真菌代谢物抑制了病原菌的繁殖(Weller et al, 2002)。大豆连作导致的大豆孢囊线虫病连续暴发多年后逐渐恢复, 形成的抑病性土壤能够抵抗孢囊线虫的侵染, 与根际和孢囊际特异富集的细菌密切相关(Hussain et al, 2018)。
当病原菌突破根际“第一道防线”进入植物内部后, 内生微生物可以通过生态位竞争、产生抗真菌化合物或增强宿主免疫来抑制病原菌, 被认为是宿主抵御病原菌入侵的“第二道防线” (Dini-Andreote, 2020)。如立枯丝核菌(Rhizoctonia solani)侵染甜菜(Beta vulgaris)根系后内生细菌如噬几丁质科和黄杆菌属显著富集, 并大量表达与真菌细胞壁降解和次级代谢产物合成相关的酶抑制病原菌繁殖(Carrión et al, 2019)。内生真菌如蜡壳耳目真菌(Serendipita vermifera)则可以通过降低病原真菌毒力因子的表达, 扩展对拟南芥和大麦等植物的保护屏障(Sarkar et al, 2019; Mahdi et al, 2022)。除了细菌和真菌类群外, 原生生物可通过直接捕食病原菌或通过生态位竞争或分泌拮抗性的代谢产物抑制病原性细菌和真菌的生长(韦中等, 2021)。如变形虫对病原青枯菌(Ralstonia solanacearum)的捕食、肉足虫对全蚀病病原真菌的捕食, 可显著降低土传病害的发病率(Xiong et al, 2020; 韦中等, 2021)。有机肥施用下捕食性原生生物还可以通过刺激香蕉(Musa Cavendish)根系微生物群落中芽孢杆菌的丰度和抑病功能来促进植物健康(Guo et al, 2022)。结合宏基因组和纯培养的研究方法发现, 被柑橘砂皮病菌(Diaporthe citri)感染的植株叶际可以通过富集具有拮抗性的泛菌属、甲基杆菌属和鞘氨醇单胞菌属菌株来抑制孢子萌发和菌丝生长(Li et al, 2022a)。但相比于根际微生物, 叶际微生物介导的植物防御相关研究尚处于起步阶段。
植物微生物介导的抑病作用机制主要有两种: (1)有益微生物通过分泌抗菌类物质或与病原菌竞争生态位和养分直接抑制病原菌的生长。如小麦穗中分离出的假单胞菌(Pseudomonas piscium ZJU60)能够大量分泌具有抑菌活性的吩嗪-1-甲酰胺(phenazine-1-carboxamide)抑制禾谷镰刀菌(Fusarium graminearum)生长和毒素合成(Chen et al, 2018)。在西瓜枯萎病病害严重暴发的土壤中, 健康西瓜植株的根际存在大量非致病性镰刀菌从而与病原性镰刀菌形成生态位竞争(Ge et al, 2021)。另如番茄根际有益菌群(如黄杆菌)可通过与病原青枯菌竞争合成糖类的前体物质, 或通过分泌抑制型铁载体与青枯菌形成铁资源竞争以抑制病原菌生长(Kwak et al, 2018; Gu et al, 2020)。(2)刺激或启动植物免疫系统间接抑制病原微生物, 即根际有益微生物通过刺激植物自身免疫系统从而增强植株整体防御能力, 也被称为诱导系统抗性(induced systemic resistance, ISR) (Pieterse et al, 2014)。如有研究发现, 将分离自健康番茄根际特异性富集的厚壁菌和放线菌重接种到土壤中会诱导植物系统抗性, 降低番茄青枯病的发生(Lee et al, 2021)。类似地, Li等(2021)研究发现, 由4个菌株构建的简单合成群落可通过低丰度细菌小陌生菌(Advenella sp.)激活植物系统性抗性和高丰度细菌抑制病原体生长的协同作用来保护植物。
2.4 提高植物对环境胁迫的适应性
植物对微生物的“呼救”策略还存在于对干旱、低温、盐碱等环境胁迫的反应中。干旱是目前全球农业生产中的主要威胁, 越来越多的研究表明植物微生物在帮助植物缓解干旱胁迫中起着重要作用(de Vries et al, 2020)。如Santos-Medellín等(2021)研究发现, 水稻受到干旱胁迫后根系大量富集的链霉菌有利于根系伸长和幼苗生长, 其促生机制可能与生长素和铁载体的产生有关。基于转录组和代谢组的研究同样发现干旱胁迫下高梁根系放线菌显著富集, 碳水化合物、氨基酸转运和代谢相关的基因高度表达(Xu et al, 2018, 2021)。在低温胁迫下, 植物内生细菌如伯克氏菌(Burkholderia)、假单胞菌等, 以及内生真菌和丛枝菌根真菌等可以通过触发早期的激素信号、提高抗氧化活性以及渗透液浓度等途径帮助植物抗寒(Acuña-Rodríguez et al, 2020)。半干旱区土地盐碱化越来越成为粮食生产的制约因子, 一些根际促生细菌、木霉菌(Trichoderma)、印度梨形孢(Serendipita indica)等可以通过分泌胞外多糖、产生脯氨酸和多胺类等渗透物质、调控植物基因表达等途径限制钠离子进入植物根系以及提高根系钾离子的吸收和钠离子的排放从而帮助植物抵御盐胁迫(Dodd & Pérez-Alfocea, 2012; Qin et al, 2016)。Schmitz等(2022)利用从沙漠植物靛蓝(Indigofera argentea)根系分离得到的5株细菌构建合成群落, 将其接种到无菌土壤中可以保护番茄免受高盐胁迫。
以上研究表明微生物组在植物养分吸收、生长发育、病害抵抗和环境胁迫适应等方面都起着重要作用, 利用植物微生物构建合成菌群在促进植物生产、减少化肥农药使用等方面具有广阔的应用前景。然而目前大部分研究还局限于实验室条件下的功能验证阶段, 其具体作用机制和在田间的应用效果还有待进一步探究。除以上有益功能外, 植物微生物组中的潜在病原菌如病原真菌、卵菌等可引起严重的植物病害, 导致生产力下降甚至植物物种的消失。如锈菌目的真菌广泛寄生于禾本科植物、裸子植物和蕨类植物上, 可以引起许多重要作物和林木病害(Helfer, 2014)。此外, 对多数植物造成危害的病原真菌(如核盘菌、炭疽菌等)也可作为其他植物的有益内生菌或共生菌存在, 即真菌的“分裂营养” (schizotrophism)现象。在一些特殊情况下, 具有分裂营养内生真菌的健康植物可能将这些菌传播到其他寄主植物上成为破坏性的病原菌, 导致寄主植物种群衰退(Tian et al, 2020)。在森林、温带草地和弃耕地等生态系统中, 病原真菌所介导的同种负密度制约假说是解释植物物种共存的重要理论(Jia et al, 2020; 刘向等, 2023)。因此, 微生物个体水平上对宿主植物表现出的性状不应被视为绝对有益或有害的, 更应该从群落水平上关注其在不同宿主植物和环境条件中产生的不同表型, 从植物-微生物-环境三者互作的角度认识植物微生物组的生态功能, 以服务于植物生产力提高、生物多样性保护和生态系统管理。
3 植物微生物群落构建机制及驱动因素
植物微生物的群落构建, 即指群落多样性形成和维持的基本生态过程, 群落生态学理论认为微生物群落构建受到扩散(如微生物的迁移)、选择(如群落受到生物和非生物因素的影响)、成种(如遗传变异)和漂变(如随机的出生和死亡事件) 4个生态进化过程的共同影响(Nemergut et al, 2013)。其中, 选择代表完全的确定性过程, 漂变代表完全的随机性过程, 而扩散和成种既包括确定性过程也包括随机性过程; 扩散和成种过程影响微生物群落多样性, 而选择和漂变会影响群落内部微生物丰度(Vellend, 2010; Zhou & Ning, 2017)。Dini-Andreote等(2015)认为土壤微生物演替过程中的初始阶段是由随机性过程控制的, 而环境的变化逐渐增加了确定性选择的重要性, 当环境稳定后, 随机性和确定性过程的相对影响也趋于稳定。但确定性和随机性过程对植物微生物群落构建的相对重要性仍不清楚, 以往大多数研究主要关注了宿主选择和环境过滤所代表的确定性过程的作用, 尤其是在农田生态系统中, 由于物种少、人为干扰强且周期较短, 其他3个生态过程(即成种、扩散和漂变)的影响相对弱。总体而言, 植物微生物群落构建的驱动因素可以概括为宿主选择、环境因子以及生物之间的互作等。
3.1 植物微生物组群落来源
大量研究表明植物微生物组有不同来源途径, 包括土壤、大气环境、邻近植物、种子和昆虫等。土壤中蕴藏着巨大的微生物多样性, 是植物微生物群落的主要来源, 为植物提供了丰富的“微生物资源库” (Vandenkoornhuyse et al, 2015)。一方面, 土壤微生物可通过被动扩散到达植物根际和根表并进一步向植物其他部位迁移(Cordovez et al, 2019)。在这个过程中, 农业活动如耕作、灌溉等人为干扰, 以及土壤动物的活动均会影响土壤微生物向植物的扩散, 如线虫对根系的取食为根内生菌的定殖打开了通道(Topalović & Heuer, 2019)。另一方面, 植物通过根系将代谢产物输入到地下, 为根际微生物提供了丰富营养物质的同时也释放了信号分子从而产生特定的选择压力, 使得植物主动招募土壤微生物(Philippot et al, 2013; Sasse et al, 2018)。最终, 部分土壤微生物通过扩散、环境过滤和宿主植物的选择成为植物内生微生物。许多研究也表明大气环境以及传粉昆虫可能是叶表微生物群落的主要来源(Humphrey & Whiteman, 2020; Gong & Xin, 2021; Xiong et al, 2021b)。此外, 来自亲代种子微生物的垂直传播也是植物微生物的一个重要来源(Shade et al, 2017; Berg & Raaijmakers, 2018)。如近期研究发现上一代橡树微生物组(细菌和真菌)可以通过种子垂直传播到下一代橡树幼苗的叶际和根系(Abdelfattah et al, 2021)。
3.2 宿主选择对植物微生物组群落构建的影响
3.2.1 植物部位生态位
植物为微生物群落提供了多种微生境, 包括根际(rhizosphere)、表生(epiphytic)和内生际(endophytic), 代表着植物不同部位生态位。在植物整株水平上, 部位生态位是决定植物微生物群落组成的主要选择压力, 即微生物组成在植物不同部位显著不同(Xiong et al, 2021a, c)。这可能归因于植物不同部位生态位的宿主免疫、功能性状及所提供的微环境各不相同(Trivedi et al, 2020)。植物根际是指根系附近受根系分泌物和氧气可利用性影响的区域。根际作为植物与土壤相互作用的界面, 被认为是一个营养丰富的微生物活动热区(Philippot et al, 2013)。植物通过根系释放大量的根际沉积物, 主要包括根系代谢产物、信号分子和死亡的根冠细胞等, 其中根系代谢产物主要包括糖类、氨基酸、有机酸等小分子物质, 为根际微生物提供了丰富的碳源(Sasse et al, 2018)。此外, 根系代谢产物中还包括一些次级代谢产物如抗菌化合物和类黄酮化合物等(Hassan & Mathesius, 2012; Zhang et al, 2017; Brunel et al, 2020)。这些代谢产物对根际微生物产生了特定的选择压力, 即根际效应(Philippot et al, 2013; Sasse et al, 2018)。与非根际土壤相比, 根际土壤微生物数量增加了几倍至几十倍(Bulgarelli et al, 2015), 变形菌门、拟杆菌门和放线菌门等类群显著富集, 与这些类群属于快速生长型的富营养型细菌, 且具有广泛利用根系碳源的能力密切相关(Vandenkoornhuyse et al, 2007; Edwards et al, 2015)。类似地, 子囊菌门(如肉座菌目)和球囊菌门(如球囊霉属(Glomus))等也能快速利用根际沉积物, 是根际环境中的优势真菌类群(Hannula et al, 2012)。与土壤微生物相比, 根系相关细菌编码了更多与趋化性、鞭毛合成、运动以及生物膜合成有关的功能基因, 根际微生物通过趋化性感知植物信号后利用鞭毛等运动器官向根系靠近, 附着在根系表面并形成生物膜(Knief et al, 2012; Levy et al, 2018)。基于分离培养的结果同样发现, 拟南芥根系分离得到的细菌拥有更多与运动相关的基因(Bai et al, 2015)。
植物表生际指根、茎、叶、花及果实等植物器官的表面环境(Hardoim et al, 2008; Philippot et al, 2013; Gong & Xin, 2021)。以叶表为例, 由于暴露在空气中面临着诸多不利条件, 如营养物质少、温湿度波动大、紫外线辐射强烈等对微生物构成较强的选择压力, 因此植物表生微生物同时受到宿主和环境的共同影响(Remus-Emsermann & Schlechter, 2018; Liu et al, 2020; Xiong et al, 2021b)。一方面, 叶际微生物可利用的营养物质非常有限, 且分布不均匀, 主要为叶片表皮细胞释放出来的碳水化合物、脂肪、有机酸和氨基酸等, 或从叶片气孔释放出来的挥发性有机物质如甲醇(van Der Wal & Leveau, 2011)。据估计, 全球植物叶片每年释放的甲醇大约有1024 g, 这为能快速消耗甲醇的甲基杆菌属细菌的定殖提供了碳源, 而鞘氨醇单胞菌属对叶表产生的碳水化合物具有高效的吸收能力, 假单胞菌属能依靠自身的运动往叶际营养丰富的地方移动, 使得这些菌群在叶际被显著富集(Remus-Emsermann & Vorholt, 2014)。另一方面, 叶际时常面临干燥和紫外线辐射等恶劣条件, 大部分叶际微生物常形成聚集体(aggregates)分布在气孔、毛状体、叶片纹理和表皮细胞连接处的凹槽内获得保护, 或通过分泌生物表面活性剂(biosurfactant)和胞外多糖类物质以抵抗养分和水分缺乏, 或产生细胞色素以抵御紫外线辐射的影响(Chang et al, 2007; Burch et al, 2014; Yoshida et al, 2017)。
相较于植物表生际, 植物内生际对微生物的选择效应更强, 这主要是归因于植物免疫系统的存在(Bulgari et al, 2014; Yao et al, 2019; Zhang et al, 2022)。通常情况下, 当微生物侵入植物内部时会释放如鞭毛蛋白、几丁质、脂多糖和延伸因子Tu衍生肽等物质, 而植物模式受体(pattern recognition receptors, PRRs)识别到微生物相关的分子模式(microbe-associated molecular patterns, MAMPs)后会启动一系列防御信号转导机制, 如产生活性氧, 激活丝裂原活化蛋白激酶(mitogen-activated protein kinase, MAPKs)等进而启动水杨酸、茉莉酸信号转导途径来进行防御(Boller & Felix, 2009; Han, 2019)。在长期进化过程中, 微生物成功进化出一系列机制以逃避或者抑制植物的免疫系统, 如通过分泌与植物蛋白结构相似的效应蛋白躲避植物受体的识别, 一些微生物可通过分泌解毒酶抑制植物活性氧的产生, 有利于其初始定殖。虽然裂解酶有助于微生物进入植物组织, 但内生菌产生的裂解酶水平通常较低从而避免了触发植物免疫反应(Levy et al, 2018; Trivedi et al, 2020)。
3.2.2 植物种类和基因型
植物遗传变异会影响植物的功能性状和免疫系统, 因此不同种类或基因型植物会特异性富集不同的微生物(Cordovez et al, 2019)。对玉米、十字花科植物和木本植物的研究均发现宿主植物物种及基因型对其根际或叶际微生物群落构建具有重要影响(Peiffer et al, 2013; Kembel et al, 2014; Wagner et al, 2016)。如芽孢杆菌属、乳杆菌科及甲基杆菌科分别是小麦、大麦及玉米的指示类群(Xiong et al, 2021c)。大尺度的田间观测也发现不同玉米和水稻基因型可通过影响根际细菌群落组成调控玉米和水稻氮素利用效率(Walters et al, 2018; Zhang et al, 2019; Yu et al, 2021)。植物种类或基因型对微生物群落组成的影响可归因于功能性状如代谢产物、根系直径、根系氮含量、比根长等因素的差异(Pérez-Jaramillo et al, 2017; Hu et al, 2018; Sweeney et al, 2021)。如甜玉米品系由于sugary1 (su1)基因突变导致胚乳蔗糖和葡萄糖浓度增加而支链淀粉含量减少, 微生物可利用碳源发生变化, 使得甜玉米根系富集更多具有固氮能力的伯克氏菌属和根瘤菌属(Rhizobium)等类群(James et al, 1995; Walters et al, 2018)。此外, 不同种类或基因型植物还可以通过影响免疫系统来调控微生物群落构建, 如有研究发现茉莉酸合成缺陷的拟南芥突变体根系细菌和古菌群落结构发生明显变化, 链霉菌属、芽孢杆菌属和肠杆菌科丰度显著增加(Carvalhais et al, 2015)。
植物驯化在农业生产中历史悠久, 在改变植物表型性状的同时也减少了植物遗传多样性进而减少了植物微生物多样性。如野生型和栽培品种的水稻种子、小麦和大麦根际均拥有差异显著的微生物群落, 且野生型植物根际微生物多样性通常更高 (Bulgarelli et al, 2015; Hassani et al, 2020; Kim et al, 2020)。与栽培作物相比, 野生植物更依赖于根际微生物帮助其吸收养分和抵抗病害, 在长期进化过程中与有益微生物形成了紧密的关系, 其根际微生物往往具有高效的养分吸收和病原菌抵御能力(Pérez-Jaramillo et al, 2018; Porter & Sachs, 2020)。因此近年来有学者提出“微生物组再野化”假说, 认为应大力加强植物微生物组学研究, 探索恢复驯化植物微生物多样性和功能的方法和策略, 以促进植物健康和生产力提高(Raaijmakers & Kiers, 2022)。
3.2.3 植物生长发育时期
植物微生物群落多样性、组成和功能会随着时间推移而改变以应对植物生长和环境变化(Müller et al, 2016)。土壤作为植物微生物组的“种子库”, 在植物生长初期根系微生物组往往与周边土壤相似, 随着植物生长发育成熟, 其选择作用增强、微生物组内的生态位竞争加剧, 植物微生物组成更加具有“植物特异性” (Chaparro et al, 2014; Copeland et al, 2015; Gao et al, 2020)。例如, 水稻根系微生物组群落结构在营养生长阶段变化较快、进入生殖生长后菌群结构趋于稳定(Edwards et al, 2018); 叶片年龄可以通过影响叶际营养物质的质量和组成进一步影响叶际微生物群落(Vorholt, 2012; Morella et al, 2020)。对玉米不同生长发育时期下不同部位生态位微生物组的研究也发现, 植物发育时期对细菌、真菌群落的alpha多样性、组成、网络互作关系有显著影响, 其中叶表的时期效应最强; 在作物发育早期有益细菌如放线菌门、伯克氏菌科和根瘤菌科等显著富集, 叶表微生物组具有更高的功能基因多样性; 在后期, 腐生真菌显著富集, 叶表微生物组中与氮同化和碳降解相关的基因显著增加(Xiong et al, 2021b)。这些研究表明植物可根据其生长发育需求选择性塑造不同的微生物群落, 代表了宿主与微生物长期共进化的结果。植物发育时期对微生物的选择效应主要与植物代谢和免疫反应密切相关, 同时还受季节性环境因素的共同影响(Durán et al, 2018; Chen et al, 2019; Xiong et al, 2021b)。
3.3 环境因子对植物微生物群落构建的影响
除植物的选择效应外, 土壤性质、气候变化、农业管理措施等非生物因素都会影响到植物微生物群落构建。土壤性质是决定根系微生物群落组成的重要因子, 如在较大的空间尺度上, 拟南芥根系细菌群落结构分异主要由土壤pH以及可溶性钙、钾、镁、铁等土壤养分因子决定(Thiergart et al, 2020)。土壤质地也可以通过影响根系构型进而影响微生物组成(Gebauer et al, 2021)。此外, 环境扰动如高强度的集约化农业管理对根际微生物群落也有影响, 与传统高强度农业集约化管理相比, 免耕或有机农业管理下小麦根部真菌网络复杂度和丛枝菌根真菌丰度显著增加(Banerjee et al, 2019)。气候变化如全球气温升高、极端气候事件等在影响植物生理和表型的同时也改变了植物微生物(Trivedi et al, 2022)。一项长期控制试验研究发现, 增温导致草本植物白花拉拉藤(Galium album)叶际gamma-变形菌、放线菌和厚壁菌丰度显著增加, 而alpha-变形菌和拟杆菌丰度降低, 这可能与高温导致气孔开度降低进而影响了叶片代谢有关(Aydogan et al, 2018)。目前的研究认为在短期内(几年到几十年), 植物对气候变化的适应主要由植物微生物组驱动, 而在长期尺度上(一个世纪到几千年), 植物微生物组与其宿主之间的生态进化可能发挥了重要的作用(Trivedi et al, 2022)。因此阐明植物微生物组对气候变化的响应及其提高植物适应性的机制, 可为预测气候变化对初级生产力的影响提供有力支撑。
3.4 生物间的相互作用对植物微生物群落构建与维持的影响
除宿主和环境因素外, 微生物之间、微生物与其他生物之间的相互作用对植物微生物组群落构建及其稳定性也具有重要影响。微生物之间可以通过营养依赖、生物膜形成、分子通讯等合作途径, 以及资源竞争、依赖竞争、捕食、抗菌化合物分泌、挥发性有机化合物释放等竞争途径来实现长期共存(Hassani et al, 2018)。如对无菌拟南芥进行根系微生物重接种试验结果发现细菌可以抑制真菌和卵菌对植物的有害影响(Durán et al, 2018)。Niu等(2017)构建了由7种细菌组成的人工合成菌群(synthetic communities, SynComs), 并通过不同组合形式将其接种到无菌玉米幼苗根部, 发现去除其中的阴沟肠杆菌(Enterobacter cloacae)后短杆菌(Curtobacterium pusillum)在群落中占据主导地位, 与单株菌相比, 合成菌群对病原真菌轮状镰刀菌(Fusarium verticillioides)有明显的抑制作用。
此外, 昆虫的取食、病原菌入侵对植物微生物群落也有重要影响。如食草昆虫通过激活植物防御机制从而重塑苦芥(Cardamine cordifolia)叶片内生细菌群落(Humphrey & Whiteman, 2020)。越来越多的研究发现病原菌入侵会显著影响植物微生物互作模式(Mendes et al, 2018; Carrión et al, 2019; Gao et al, 2021; Ge et al, 2021)。镰刀菌属(Fusarium)病原真菌入侵引起的西瓜、辣椒和大豆枯萎病导致植物微生物互作网络发生显著改变, 如尖孢镰刀菌西瓜专化型(Fusarium oxysporum f. sp. niveum)入侵导致西瓜根际细菌-真菌互作网络复杂度降低(Mendes et al, 2018; Gao et al, 2021; Ge et al, 2021)。虽然人们已认识到微生物之间、微生物与其他生物成员之间的相互作用对植物微生物组组成和稳定具有不可忽视的作用, 但目前对相互作用的机制仍知之甚少。
综上所述, 宿主选择、环境因子和生物互作共同驱动了植物微生物群落构建, 从土壤到植物表面再到内部, 宿主选择的效应逐渐增强, 而环境因子的影响逐渐减弱, 植物表面(如根表和叶表)微生物受到宿主选择和环境效应共同影响(图2)。系统研究植物微生物组的群落构建机制, 阐明不同生态系统中植物微生物的来源, 宿主植物、环境和人为干扰等如何塑造微生物组的结构和功能, 以及高度多样的植物微生物组如何响应复杂的环境条件并影响宿主植物的生长, 对未来植物微生物组精准调控和生态系统可持续管理具有重要意义。
| Box 1 植物微生物的主要生态功能及其作用机制 | |||
|---|---|---|---|
| 主要功能 | 微生物类群 | 作用机制 | 参考文献 |
| 调节植物生长发育 | 恶臭假单胞菌 Pseudomonas putida | 降低植物乙烯水平促进生长的同时引发强烈的胁迫过敏反应影响植物生长-防御权衡 | Ravanbakhsh et al, 2019 |
| 贪噬菌属 Variovorax | 拥有高度保守的生长素(IAA)降解操纵子, 通过降解植物生长素来解除根系生长抑制 | Finkel et al, 2020 | |
| 红球菌属、黄杆菌属 Rhodococcus, Flavobacterium | 直接产生IAA和铁载体促进根系伸长 | Belimov et al, 2005 | |
| 促进植物养分吸收 | 根瘤菌属、甲基杆菌属、克雷伯氏菌属Rhizobium, Methylobacterium, Klebsiella | 固定空气中的氮气, 为植物提供氮素营养 | Richardson et al, 2009; Zhang et al, 2020; Madhaiyan et al, 2015; Zhang et al, 2022 |
| 丛枝菌根真菌 Arbuscular mycorrhizal fungi | 产生有机酸或铁载体将土壤中难溶组分氧化、增溶或螯合成植物可利用的养分 | van der Heijden et al, 2017 | |
| 猴假单胞菌 Pseudomonas simiae | 间接诱导植物基因表达和代谢产物分泌以促进养分吸收 | Voges et al, 2019; Harbort et al, 2020 | |
| 提高植物抗病性 | 绿针假单胞菌双鱼亚种 Pseudomonas piscium | 直接分泌抑菌物质如抗生素、溶菌酶等 | Chen et al, 2018 |
| 非致病镰刀菌(如轮枝镰刀菌 Fusarium verticillioides和腐皮镰刀菌 Fusarium solani) | 直接与病原菌生态位竞争 | Ge et al, 2021 | |
| 黄杆菌属 Flavobacterium | 直接与病原菌养分竞争 | Kwak et al, 2018 | |
| 厚壁菌门 Firmicutes 放线菌门 Actinobacteria | 间接诱导植物系统抗性 | Lee et al, 2021 | |
| 提高植物环境胁迫适应性 | 链霉菌属 Streptomyces | 产生IAA和铁载体以促进根系伸长抵御干旱胁迫 | Santos-Medellín et al, 2021 |
| 伯克氏菌属、假单胞菌属、丛枝菌根真菌 Burkholderia, Pseudomonas, Arbuscular mycorrhizal fungi | 触发植物早期激素信号、提高抗氧化活性以及渗透液浓度等途径帮助植物抗寒 | Acuña-Rodríguez et al, 2020 | |
| 木霉菌、印度梨形孢 Trichoderma, Serendipita indica | 分泌胞外多糖结合过量钠离子并限制钠离子进入根系、产生渗透物质、调控植物基因表达抵御盐胁迫 | Dodd & Pérez-Alfocea, 2012; Qin et al, 2016 | |
图2
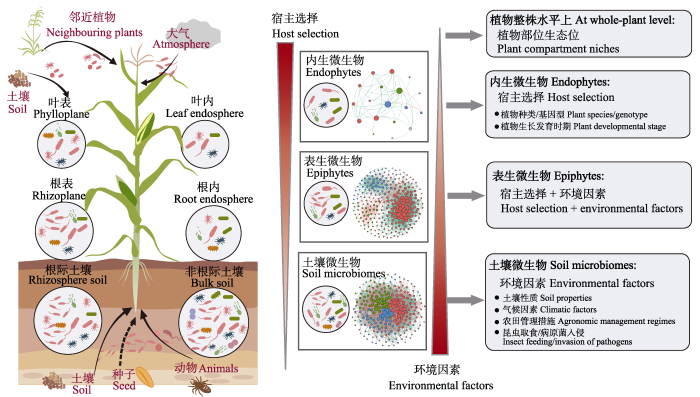
图2
植物微生物组来源以及宿主选择和环境因素共同驱动的土壤-植物连续体微生物群落构建模型
Fig. 2
Sources of plant microbiomes and a conceptual model for microbial community assembly on the soil-plant continuum driven by the interactive effect of host selection and environmental effect
4 展望
综上所述, 得益于多学科的融合和多组学技术的发展, 近十多年来对植物微生物群落多样性及组成特征、生态功能、驱动因素, 以及植物-微生物相互作用机制等都有了初步的认识, 但距离精准调控微生物组服务于植物生产力提高和生态系统健康维持还有很长的距离, 因此, 在未来的研究中, 还有以下方面需要深入探讨:
(1)核心微生物组挖掘和合成群落应用。在多种因素的共同影响下, 植物塑造了多样化的微生物群落, 研究人员将在多种宿主植物微生物群落中普遍存在且发挥关键作用的微生物类群称为“核心微生物组” (core microbiota) (Trivedi et al, 2020)。核心微生物组的存在降低了植物微生物组的复杂性, 为在可控实验体系下构建合成群落提供了理论依据和实践价值。但是目前核心微生物组的识别主要基于微生物互作网络和机器学习等分析手段, 一方面不足以解释物种间的互作, 也不能表明因果关系, 另一方面对关键物种的生理生态特征及其作用机制尚不清楚。因此未来的研究需要结合高通量培养组学技术验证核心微生物组的功能特征, 进一步利用合成群落验证其实际功能及作用机制, 为未来植物微生物组精准调控提供策略和方案。
(2)植物-微生物互作的分子调控机制。宿主遗传是植物微生物群落构建的重要驱动因素, 然而, 确定控制特定微生物定殖的植物遗传位点仍然存在较大挑战。全基因组关联研究(genome wide association studies, GWASs)可以将微生物组和影响其定殖的宿主基因位点、宿主表型联系起来, 但相关的研究进展还较少(He et al, 2021)。因此未来的研究中需要对有利于特定有益微生物定殖的植物基因给予更多的关注, 并在作物育种过程中对这些基因进行筛选, 通过宿主修饰促进有益微生物群落的建立。此外, 有研究表明种子中微生物可垂直传播给下一代植物, 未来需要进一步探索微生物从种子传播到植物根、茎、叶的特征和效率, 以期通过传递植物有益微生物群系的方法为植物繁育开辟新途径。
(3)植物微生物群落水平上的互作机制。健康的植物组织中定殖着种类繁多的微生物, 这些微生物成员之间已经进化出极其复杂的相互作用关系, 但是很多研究关注的是物种水平, 在群落水平上的研究还处于起步阶段。因此未来需要基于宏基因组、转录组、蛋白组、代谢组、培养组等多组学手段综合分析, 以期从群落层面全面探究植物微生物成员之间的相互作用对植物微生物群落稳定性以及宿主健康的重要作用。
参考文献
Experimental evidence of microbial inheritance in plants and transmission routes from seed to phyllosphere and root
DOI:10.1111/1462-2920.15392
PMID:33427409
[本文引用: 1]

While the environment is considered the primary origin of the plant microbiome, the potential role of seeds as a source of transmitting microorganisms has not received much attention. Here we tested the hypothesis that the plant microbiome is partially inherited through vertical transmission. An experimental culturing device was constructed to grow oak seedlings in a microbe-free environment while keeping belowground and aboveground tissues separated. The microbial communities associated with the acorn's embryo and pericarp and the developing seeding's phyllosphere and root systems were analysed using amplicon sequencing of fungal ITS and bacterial 16S rDNA. Results showed that the seed microbiome is diverse and non-randomly distributed within an acorn. The microbial composition of the phyllosphere was diverse and strongly resembled the composition found in the embryo, whereas the roots and pericarp each had a less diverse and distinct microbial community. Our findings demonstrate a high level of microbial diversity and spatial partitioning of the fungal and bacterial community within both seed and seedling, indicating inheritance, niche differentiation and divergent transmission routes for the establishment of root and phyllosphere communities.© 2021 The Authors. Environmental Microbiology published by Society for Applied Microbiology and John Wiley & Sons Ltd.
Functional roles of microbial symbionts in plant cold tolerance
DOI:10.1111/ele.13502
PMID:32281227
[本文引用: 2]
In this review, we examine the functional roles of microbial symbionts in plant tolerance to cold and freezing stresses. The impacts of symbionts on antioxidant activity, hormonal signaling and host osmotic balance are described, including the effects of the bacterial endosymbionts Burkholderia, Pseudomonas and Azospirillum on photosynthesis and the accumulation of carbohydrates such as trehalose and raffinose that improve cell osmotic regulation and plasma membrane integrity. The influence of root fungal endophytes and arbuscular mycorrhizal fungi on plant physiology at low temperatures, for example their effects on nutrient acquisition and the accumulation of indole-3-acetic acid and antioxidants in tissues, are also reviewed. Meta-analyses are presented showing that aspects of plant performance (shoot biomass, relative water content, sugar and proline concentrations and F /F ) are enhanced in symbiotic plants at low (-1 to 15 °C), but not at high (20-26 °C), temperatures. We discuss the implications of microbial symbionts for plant performance at low and sub-zero temperatures in the natural environment and propose future directions for research into the effects of symbionts on the cold and freezing tolerances of plants, concluding that further studies should routinely incorporate symbiotic microbes in their experimental designs.© 2020 John Wiley & Sons Ltd/CNRS.
Root-associated fungal microbiota of nonmycorrhizal Arabis alpina and its contribution to plant phosphorus nutrition
Long-term warming shifts the composition of bacterial communities in the phyllosphere of Galium album in a permanent grassland field-experiment
DOI:10.3389/fmicb.2018.00144
PMID:29487575
[本文引用: 1]
Global warming is currently a much discussed topic with as yet largely unexplored consequences for agro-ecosystems. Little is known about the warming effect on the bacterial microbiota inhabiting the plant surface (phyllosphere), which can have a strong impact on plant growth and health, as well as on plant diseases and colonization by human pathogens. The aim of this study was to investigate the effect of moderate surface warming on the diversity and composition of the bacterial leaf microbiota of the herbaceous plant Galium album. Leaves were collected from four control and four surface warmed (+2 degrees C) plots located at the field site of the Environmental Monitoring and Climate Impact Research Station Linden in Germany over a 6-year period. Warming had no effect on the concentration of total number of cells attached to the leaf surface as counted by Sybr Green I staining after detachment, but changes in the diversity and phylogenetic composition of the bacterial leaf microbiota analyzed by bacterial 16S rRNA gene Illumina amplicon sequencing were observed. The bacterial phyllosphere microbiota were dominated by Proteobacteria, Bacteroidetes, and Actinobacteria. Warming caused a significant higher relative abundance of members of the Gammaproteobacteria, Actinobacteria, and Firmicutes, and a lower relative abundance of members of the Alphaproteobacteria and Bacteroidetes. Plant beneficial bacteria like Sphingomonas spp. and Rhizobium spp. occurred in significantly lower relative abundance in leaf samples of warmed plots. In contrast, several members of the Enterobacteriaceae, especially Enterobacter and Erwinia, and other potential plant or human pathogenic genera such as Acinetobacter and insect-associated Buchnera and Wolbachia spp. occurred in higher relative abundances in the phyllosphere samples from warmed plots. This study showed for the first time the long-term impact of moderate (+2 degrees C) surface warming on the phyllosphere microbiota on plants. A reduction of beneficial bacteria and an enhancement of potential pathogenic bacteria in the phyllosphere of plants may indicate that this aspect of the ecosystem which has been largely neglected up till now, can be a potential risk for pathogen transmission in agro-ecosystems in the near future.
The root microbiome: Community assembly and its contributions to plant fitness
DOI:10.1111/jipb.13226
[本文引用: 1]
The root microbiome refers to the community of microbes living in association with a plant's roots, and includes mutualists, pathogens, and commensals. Here we focus on recent advances in the study of root commensal community which is the major research object of microbiome-related researches. With the rapid development of new technologies, plant–commensal interactions can be explored with unprecedented breadth and depth. Both the soil environment and the host plant drive commensal community assembly. The bulk soil is the seed bank of potential commensals, and plants use root exudates and immune responses to build healthy microbial communities from the available microbes. The plant microbiome extends the functional system of plants by participating in a variety of processes, including nutrient absorption, growth promotion, and resistance to biotic and abiotic stresses. Plants and their microbiomes have evolved adaptation strategies over time. However, there is still a huge gap in our understanding of the regulatory mechanisms of plant–commensal interactions. In this review, we summarize recent research on the assembly of root microbial communities and the effects of these communities on plant growth and development, and look at the prospects for promoting sustainable agricultural development through the study of the root microbiome.
Functional overlap of the Arabidopsis leaf and root microbiota
Agricultural intensification reduces microbial network complexity and the abundance of keystone taxa in roots
DOI:10.1038/s41396-019-0383-2
[本文引用: 1]
Root-associated microbes play a key role in plant performance and productivity, making them important players in agroecosystems. So far, very few studies have assessed the impact of different farming systems on the root microbiota and it is still unclear whether agricultural intensification influences the structure and complexity of microbial communities. We investigated the impact of conventional, no-till, and organic farming on wheat root fungal communities using PacBio SMRT sequencing on samples collected from 60 farmlands in Switzerland. Organic farming harbored a much more complex fungal network with significantly higher connectivity than conventional and no-till farming systems. The abundance of keystone taxa was the highest under organic farming where agricultural intensification was the lowest. We also found a strong negative association (R2 = 0.366; P < 0.0001) between agricultural intensification and root fungal network connectivity. The occurrence of keystone taxa was best explained by soil phosphorus levels, bulk density, pH, and mycorrhizal colonization. The majority of keystone taxa are known to form arbuscular mycorrhizal associations with plants and belong to the orders Glomerales, Paraglomerales, and Diversisporales. Supporting this, the abundance of mycorrhizal fungi in roots and soils was also significantly higher under organic farming. To our knowledge, this is the first study to report mycorrhizal keystone taxa for agroecosystems, and we demonstrate that agricultural intensification reduces network complexity and the abundance of keystone taxa in the root microbiome.
Cadmium-tolerant plant growth-promoting bacteria associated with the roots of Indian mustard (Brassica juncea L. Czern.)
DOI:10.1016/j.soilbio.2004.07.033 URL [本文引用: 2]
The rhizosphere microbiome and plant health
DOI:10.1016/j.tplants.2012.04.001
PMID:22564542
[本文引用: 1]
The diversity of microbes associated with plant roots is enormous, in the order of tens of thousands of species. This complex plant-associated microbial community, also referred to as the second genome of the plant, is crucial for plant health. Recent advances in plant-microbe interactions research revealed that plants are able to shape their rhizosphere microbiome, as evidenced by the fact that different plant species host specific microbial communities when grown on the same soil. In this review, we discuss evidence that upon pathogen or insect attack, plants are able to recruit protective microorganisms, and enhance microbial activity to suppress pathogens in the rhizosphere. A comprehensive understanding of the mechanisms that govern selection and activity of microbial communities by plant roots will provide new opportunities to increase crop production.Copyright © 2012 Elsevier Ltd. All rights reserved.
Saving seed microbiomes
DOI:10.1038/s41396-017-0028-2 [本文引用: 1]
A renaissance of elicitors: Perception of microbe-associated molecular patterns and danger signals by pattern-recognition receptors
DOI:10.1146/annurev.arplant.57.032905.105346
PMID:19400727
[本文引用: 1]
Microbe-associated molecular patterns (MAMPs) are molecular signatures typical of whole classes of microbes, and their recognition plays a key role in innate immunity. Endogenous elicitors are similarly recognized as damage-associated molecular patterns (DAMPs). This review focuses on the diversity of MAMPs/DAMPs and on progress to identify the corresponding pattern recognition receptors (PRRs) in plants. The two best-characterized MAMP/PRR pairs, flagellin/FLS2 and EF-Tu/EFR, are discussed in detail and put into a phylogenetic perspective. Both FLS2 and EFR are leucine-rich repeat receptor kinases (LRR-RKs). Upon treatment with flagellin, FLS2 forms a heteromeric complex with BAK1, an LRR-RK that also acts as coreceptor for the brassinolide receptor BRI1. The importance of MAMP/PRR signaling for plant immunity is highlighted by the finding that plant pathogens use effectors to inhibit PRR complexes or downstream signaling events. Current evidence indicates that MAMPs, DAMPs, and effectors are all perceived as danger signals and induce a stereotypic defense response.
Towards unraveling macroecological patterns in rhizosphere microbiomes
DOI:S1360-1385(20)30148-5
PMID:32467065
[本文引用: 1]
It is generally accepted that plants locally influence the composition and activity of their rhizosphere microbiome, and that rhizosphere community assembly further involves a hierarchy of constraints with varying strengths across spatial and temporal scales. However, our knowledge of rhizosphere microbiomes is largely based on single-location and time-point studies. Consequently, it remains difficult to predict patterns at large landscape scales, and we lack a clear understanding of how the rhizosphere microbiome forms and is maintained by drivers beyond the influence of the plant. By synthesizing recent literature and collating data on rhizosphere microbiomes, we point out the opportunities and challenges offered by advances in molecular biology, bioinformatics, and data availability. Specifically, we highlight the use of exact sequence variants, coupled with existing and newly generated data to decipher the rules of rhizosphere community assembly across large spatial and taxonomic scales.Copyright © 2020 The Authors. Published by Elsevier Ltd.. All rights reserved.
Structure and function of the bacterial root microbiota in wild and domesticated barley
Endophytic bacterial community of grapevine leaves influenced by sampling date and phytoplasma infection process
DOI:10.1186/1471-2180-14-1 [本文引用: 1]
The hygroscopic biosurfactant syringafactin produced by Pseudomonas syringae enhances fitness on leaf surfaces during fluctuating humidity
DOI:10.1111/emi.2014.16.issue-7 URL [本文引用: 1]
Pathogen-induced activation of disease-suppressive functions in the endophytic root microbiome
DOI:10.1126/science.aaw9285
PMID:31672892
[本文引用: 3]
Microorganisms living inside plants can promote plant growth and health, but their genomic and functional diversity remain largely elusive. Here, metagenomics and network inference show that fungal infection of plant roots enriched for Chitinophagaceae and Flavobacteriaceae in the root endosphere and for chitinase genes and various unknown biosynthetic gene clusters encoding the production of nonribosomal peptide synthetases (NRPSs) and polyketide synthases (PKSs). After strain-level genome reconstruction, a consortium of and was designed that consistently suppressed fungal root disease. Site-directed mutagenesis then revealed that a previously unidentified NRPS-PKS gene cluster from was essential for disease suppression by the endophytic consortium. Our results highlight that endophytic root microbiomes harbor a wealth of as yet unknown functional traits that, in concert, can protect the plant inside out.Copyright © 2019 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. No claim to original U.S. Government Works.
Linking jasmonic acid signaling, root exudates, and rhizosphere microbiomes
DOI:10.1094/MPMI-01-15-0016-R
PMID:26035128
[本文引用: 1]
Jasmonic acid (JA) is an essential hormone in plant development and defense responses in Arabidopsis thaliana. Exogenous treatment with JA has recently been shown to alter root exudate profiles and the composition of root-associated bacterial communities. However, it is currently unknown whether disruptions of the JA in the rhizosphere affect root exudation profiles and the relative abundance of bacteria and archaea in the rhizosphere. In the present study, two Arabidopsis mutants that are disrupted in different branches of the jasmonate pathway, namely myc2 and med25, were cultivated in nutrient solution and soil to profile root exudates and bacterial and archaeal communities, respectively. Compared with the wild type, both mutants showed distinct exudation patterns, including lower amounts of asparagine, ornithine, and tryptophan, as well as distinct bacterial and archaeal community composition, as illustrated by an increased abundance of Streptomyces, Bacillus, and Lysinibacillus taxa in the med25 rhizosphere and an Enterobacteriaceae population in myc2. Alternatively, the Clostridiales population was less abundant in the rhizosphere of both mutants. Similarities between plant genotypes were highly correlated, as determined by operational taxonomic units in the rhizosphere and metabolites in root exudates. This strongly suggests that root exudates play a major role in modulating changes in microbial community composition upon plant defense responses.
Protist diversity and community complexity in the rhizosphere of switchgrass are dynamic as plants develop
DOI:10.1186/s40168-021-01042-9
PMID:33910643
[本文引用: 1]
Despite their widespread distribution and ecological importance, protists remain one of the least understood components of the soil and rhizosphere microbiome. Knowledge of the roles that protists play in stimulating organic matter decomposition and shaping microbiome dynamics continues to grow, but there remains a need to understand the extent to which biological and environmental factors mediate protist community assembly and dynamics. We hypothesize that protists communities are filtered by the influence of plants on their rhizosphere biological and physicochemical environment, resulting in patterns of protist diversity and composition that mirror previously observed diversity and successional dynamics in rhizosphere bacterial communities.We analyzed protist communities associated with the rhizosphere and bulk soil of switchgrass (SG) plants (Panicum virgatum) at different phenological stages, grown in two marginal soils as part of a large-scale field experiment. Our results reveal that the diversity of protists is lower in rhizosphere than bulk soils, and that temporal variations depend on soil properties but are less pronounced in rhizosphere soil. Patterns of significantly prevalent protists groups in the rhizosphere suggest that most protists play varied ecological roles across plant growth stages and that some plant pathogenic protists and protists with omnivorous diets reoccur over time in the rhizosphere. We found that protist co-occurrence network dynamics are more complex in the rhizosphere compared to bulk soil. A phylogenetic bin-based null model analysis showed that protists' community assembly in our study sites is mainly controlled by homogenous selection and dispersal limitation, with stronger selection in rhizosphere than bulk soil as SG grew and senesced.We demonstrate that environmental filtering is a dominant determinant of overall protist community properties and that at the rhizosphere level, plant control on the physical and biological environment is a critical driver of protist community composition and dynamics. Since protists are key contributors to plant nutrient availability and bacterial community composition and abundance, mapping and understanding their patterns in rhizosphere soil is foundational to understanding the ecology of the root-microbe-soil system. Video Abstract.
Alginate production by Pseudomonas putida creates a hydrated microenvironment and contributes to biofilm architecture and stress tolerance under water-limiting conditions
DOI:10.1128/JB.00727-07
URL
[本文引用: 1]
\n Biofilms exist in a variety of habitats that are routinely or periodically not saturated with water, and residents must integrate cues on water abundance (matric stress) or osmolarity (solute stress) into lifestyle strategies. Here we examine this hypothesis by assessing the extent to which alginate production by\n Pseudomonas putida\n strain mt-2 and by other fluorescent pseudomonads occurs in response to water limitations and how the presence of alginate in turn influences biofilm development and stress tolerance. Total exopolysaccharide (EPS) and alginate production increased with increasing matric, but not solute, stress severity, and alginate was a significant component, but not the major component, of EPS. Alginate influenced biofilm architecture, resulting in biofilms that were taller, covered less surface area, and had a thicker EPS layer at the air interface than those formed by an mt-2\n algD\n mutant under water-limiting conditions, properties that could contribute to less evaporative water loss. We examined this possibility and show that alginate reduces the extent of water loss from biofilm residents by using a biosensor to quantify the water potential of individual cells and by measuring the extent of dehydration-mediated changes in fatty acid composition following a matric or solute stress shock. Alginate deficiency decreased survival of desiccation not only by\n P. putida\n but also by\n Pseudomonas aeruginosa\n PAO1 and\n Pseudomonas syringae\n pv. syringae B728a. Our findings suggest that in response to water-limiting conditions, pseudomonads produce alginate, which influences biofilm development and EPS physiochemical properties. Collectively these responses may facilitate the maintenance of a hydrated microenvironment, protecting residents from desiccation stress and increasing survival.\n
Rhizosphere microbiome assemblage is affected by plant development
DOI:10.1038/ismej.2013.196 [本文引用: 1]
Root-associated microbiomes of wheat under the combined effect of plant development and nitrogen fertilization
DOI:10.1186/s40168-019-0750-2
PMID:31640813
[本文引用: 1]
Plant roots assemble microbial communities both inside the roots and in the rhizosphere, and these root-associated microbiomes play pivotal roles in plant nutrition and productivity. Although it is known that increased synthetic fertilizer input in Chinese farmlands over the past 50 years has resulted in not only increased yields but also environmental problems, we lack a comprehensive understanding of how crops under elevated nutrient input shape root-associated microbial communities, especially through adjusting the quantities and compositions of root metabolites and exudates.The compositions of bacterial and fungal communities from the roots and rhizosphere of wheat (Triticum aestivum L.) under four levels of long-term inorganic nitrogen (N) fertilization were characterized at the tillering, jointing and ripening stages. The root-released organic carbon (ROC), organic acids in the root exudates and soil organic carbon (SOC) and soil active carbon (SAC) in the rhizosphere were quantified.ROC levels varied dramatically across wheat growth stages and correlated more with the bacterial community than with the fungal community. Rhizosphere SOC and SAC levels were elevated by long-term N fertilization but varied only slightly across growth stages. Variation in the microbial community structure across plant growth stages showed a decreasing trend with N fertilization level in the rhizosphere. In addition, more bacterial and fungal genera were significantly correlated in the jointing and ripening stages than in the tillering stage in the root samples. A number of bacterial genera that shifted in response to N fertilization, including Arthrobacter, Bacillus and Devosia, correlated significantly with acetic acid, oxalic acid, succinic acid and tartaric acid levels.Our results indicate that both plant growth status and N input drive changes in the microbial community structure in the root zone of wheat. Plant growth stage demostrated a stronger influence on bacterial than on fungal community composition. A number of bacterial genera that have been described as plant growth-promoting rhizobacteria (PGPR) responded positively to N fertilization, and their abundance correlated significantly with the organic acid level, suggesting that the secretion of organic acids may be a strategy developed by plants to recruit beneficial microbes in the root zone to cope with high N input. These results provide novel insight into the associations among increased N input, altered carbon availability, and shifts in microbial communities in the plant roots and rhizosphere of intensive agricultural ecosystems.
Wheat microbiome bacteria can reduce virulence of a plant pathogenic fungus by altering histone acetylation
DOI:10.1038/s41467-018-05683-7
PMID:30143616
[本文引用: 2]
Interactions between bacteria and fungi have great environmental, medical, and agricultural importance, but the molecular mechanisms are largely unknown. Here, we study the interactions between the bacterium Pseudomonas piscium, from the wheat head microbiome, and the plant pathogenic fungus Fusarium graminearum. We show that a compound secreted by the bacteria (phenazine-1-carboxamide) directly affects the activity of fungal protein FgGcn5, a histone acetyltransferase of the SAGA complex. This leads to deregulation of histone acetylation at H2BK11, H3K14, H3K18, and H3K27 in F. graminearum, as well as suppression of fungal growth, virulence, and mycotoxin biosynthesis. Therefore, an antagonistic bacterium can inhibit growth and virulence of a plant pathogenic fungus by manipulating fungal histone modification.
Seasonal community succession of the phyllosphere microbiome
DOI:10.1094/MPMI-10-14-0331-FI
PMID:25679538
[本文引用: 1]
The leaf microbiome is influenced by both biotic and abiotic factors. Currently, we know little about the relative importance of these factors in determining microbiota composition and dynamics. To explore this issue, we collected weekly leaf samples over a 98-day growing season from multiple cultivars of common bean, soybean, and canola planted at three locations in Ontario, Canada, and performed Illumina-based microbiome analysis. We find that the leaf microbiota at the beginning of the season is very strongly influenced by the soil microbiota but, as the season progresses, it differentiates, becomes significantly less diverse, and transitions to having a greater proportion of leaf-specific taxa that are shared among all samples. A phylogenetic investigation of communities by reconstruction of unobserved states imputation of microbiome function inferred from the taxonomic data found significant differences between the soil and leaf microbiome, with a significant enrichment of motility gene categories in the former and metabolic gene categories in the latter. A network co-occurrence analysis identified two highly connected clusters as well as subclusters of putative pathogens and growth-promoting bacteria. These data reveal some of the complex ecological dynamics that occur in microbial communities over the course of a growing season and highlight the importance of community succession.
Ecology and evolution of plant microbiomes
DOI:10.1146/annurev-micro-090817-062524
PMID:31091418
[本文引用: 2]
Microorganisms colonizing plant surfaces and internal tissues provide a number of life-support functions for their host. Despite increasing recognition of the vast functional capabilities of the plant microbiome, our understanding of the ecology and evolution of the taxonomically hyperdiverse microbial communities is limited. Here, we review current knowledge of plant genotypic and phenotypic traits as well as allogenic and autogenic factors that shape microbiome composition and functions. We give specific emphasis to the impact of plant domestication on microbiome assembly and how insights into microbiomes of wild plant relatives and native habitats can contribute to reinstate or enrich for microorganisms with beneficial effects on plant growth, development, and health. Finally, we introduce new concepts and perspectives in plant microbiome research, in particular how community ecology theory can provide a mechanistic framework to unravel the interplay of distinct ecological processes-i.e., selection, dispersal, drift, diversification-that structure the plant microbiome.
Harnessing rhizosphere microbiomes for drought- resilient crop production
DOI:10.1126/science.aaz5192
URL
[本文引用: 2]
Root-associated microbes can improve plant growth, and they offer the potential to increase crop resilience to future drought. Although our understanding of the complex feedbacks between plant and microbial responses to drought is advancing, most of our knowledge comes from non-crop plants in controlled experiments. We propose that future research efforts should attempt to quantify relationships between plant and microbial traits, explicitly focus on food crops, and include longer-term experiments under field conditions. Overall, we highlight the need for improved mechanistic understanding of the complex feedbacks between plants and microbes during, and particularly after, drought. This requires integrating ecology with plant, microbiome, and molecular approaches and is central to making crop production more resilient to our future climate.
A global atlas of the dominant bacteria found in soil
Endophytes: The second layer of plant defense
DOI:S1360-1385(20)30024-8
PMID:32191867
[本文引用: 1]
Microorganisms in association with roots can protect plants against soil-borne diseases. A recent study mechanistically revealed how root endophytes act as a second microbiological layer of plant defense. Integrating ecological concepts with principles of plant pathology provides an innovative way to manipulate and engineer beneficial plant microbiomes.Copyright © 2020 Elsevier Ltd. All rights reserved.
Disentangling mechanisms that mediate the balance between stochastic and deterministic processes in microbial succession
Microbial amelioration of crop salinity stress
DOI:10.1093/jxb/ers033
PMID:22403432
[本文引用: 2]
The use of soil and irrigation water with a high content of soluble salts is a major limiting factor for crop productivity in the semi-arid areas of the world. While important physiological insights about the mechanisms of salt tolerance in plants have been gained, the transfer of such knowledge into crop improvement has been limited. The identification and exploitation of soil microorganisms (especially rhizosphere bacteria and mycorrhizal fungi) that interact with plants by alleviating stress opens new alternatives for a pyramiding strategy against salinity, as well as new approaches to discover new mechanisms involved in stress tolerance. Although these mechanisms are not always well understood, beneficial physiological effects include improved nutrient and water uptake, growth promotion, and alteration of plant hormonal status and metabolism. This review aims to evaluate the beneficial effects of soil biota on the plant response to saline stress, with special reference to phytohormonal signalling mechanisms that interact with key physiological processes to improve plant tolerance to the osmotic and toxic components of salinity. Improved plant nutrition is a quite general beneficial effect and may contribute to the maintenance of homeostasis of toxic ions under saline stress. Furthermore, alteration of crop hormonal status to decrease evolution of the growth-retarding and senescence-inducing hormone ethylene (or its precursor 1-aminocyclopropane-1-carboxylic acid), or to maintain source-sink relations, photosynthesis, and biomass production and allocation (by altering indole-3-acetic acid and cytokinin biosynthesis) seem to be promising target processes for soil biota-improved crop salt tolerance.
Microbial interkingdom interactions in roots promote Arabidopsis survival
DOI:10.1016/j.cell.2018.10.020 URL [本文引用: 2]
Structure, variation, and assembly of the root-associated microbiomes of rice
Compositional shifts in root-associated bacterial and archaeal microbiota track the plant life cycle in field-grown rice
DOI:10.1371/journal.pbio.2003862 URL [本文引用: 1]
A few Ascomycota taxa dominate soil fungal communities worldwide
DOI:10.1038/s41467-019-10373-z
PMID:31147554
[本文引用: 1]
Despite having key functions in terrestrial ecosystems, information on the dominant soil fungi and their ecological preferences at the global scale is lacking. To fill this knowledge gap, we surveyed 235 soils from across the globe. Our findings indicate that 83 phylotypes (<0.1% of the retrieved fungi), mostly belonging to wind dispersed, generalist Ascomycota, dominate soils globally. We identify patterns and ecological drivers of dominant soil fungal taxa occurrence, and present a map of their distribution in soils worldwide. Whole-genome comparisons with less dominant, generalist fungi point at a significantly higher number of genes related to stress-tolerance and resource uptake in the dominant fungi, suggesting that they might be better in colonising a wide range of environments. Our findings constitute a major advance in our understanding of the ecology of fungi, and have implications for the development of strategies to preserve them and the ecosystem functions they provide.
A single bacterial genus maintains root growth in a complex microbiome
DOI:10.1038/s41586-020-2778-7 [本文引用: 2]
Fungal community assembly in drought-stressed sorghum shows stochasticity, selection, and universal ecological dynamics
DOI:10.1038/s41467-019-13913-9
PMID:31911594
[本文引用: 1]
Community assembly of crop-associated fungi is thought to be strongly influenced by deterministic selection exerted by the plant host, rather than stochastic processes. Here we use a simple, sorghum system with abundant sampling to show that stochastic forces (drift or stochastic dispersal) act on fungal community assembly in leaves and roots early in host development and when sorghum is drought stressed, conditions when mycobiomes are small. Unexpectedly, we find no signal for stochasticity when drought stress is relieved, likely due to renewed selection by the host. In our experimental system, the host compartment exerts the strongest effects on mycobiome assembly, followed by the timing of plant development and lastly by plant genotype. Using a dissimilarity-overlap approach, we find a universality in the forces of community assembly of the mycobiomes of the different sorghum compartments and in functional guilds of fungi.
Disease-induced changes in plant microbiome assembly and functional adaptation
DOI:10.1186/s40168-021-01138-2
PMID:34526096
[本文引用: 4]
The plant microbiome is an integral part of the host and increasingly recognized as playing fundamental roles in plant growth and health. Increasing evidence indicates that plant rhizosphere recruits beneficial microbes to the plant to suppress soil-borne pathogens. However, the ecological processes that govern plant microbiome assembly and functions in the below- and aboveground compartments under pathogen invasion are not fully understood. Here, we studied the bacterial and fungal communities associated with 12 compartments (e.g., soils, roots, stems, and fruits) of chili pepper (Capsicum annuum L.) using amplicons (16S and ITS) and metagenomics approaches at the main pepper production sites in China and investigated how Fusarium wilt disease (FWD) affects the assembly, co-occurrence patterns, and ecological functions of plant-associated microbiomes.The amplicon data analyses revealed that FWD affected less on the microbiome of pepper reproductive organs (fruit) than vegetative organs (root and stem), with the strongest impact on the upper stem epidermis. Fungal intra-kingdom networks were less stable and their communities were more sensitive to FWD than the bacterial communities. The analysis of microbial interkingdom network further indicated that FWD destabilized the network and induced the ecological importance of fungal taxa. Although the diseased plants were more susceptible to colonization by other pathogenic fungi, their below- and aboveground compartments can also recruit potential beneficial bacteria. Some of the beneficial bacterial taxa enriched in the diseased plants were also identified as core taxa for plant microbiomes and hub taxa in networks. On the other hand, metagenomic analysis revealed significant enrichment of several functional genes involved in detoxification, biofilm formation, and plant-microbiome signaling pathways (i.e., chemotaxis) in the diseased plants.Together, we demonstrate that a diseased plant could recruit beneficial bacteria and mitigate the changes in reproductive organ microbiome to facilitate host or its offspring survival. The host plants may attract the beneficial microbes through the modulation of plant-microbiome signaling pathways. These findings significantly advance our understanding on plant-microbiome interactions and could provide fundamental and important data for harnessing the plant microbiome in sustainable agriculture. Video abstract.© 2021. The Author(s).
Rootstock rescues watermelon from Fusarium wilt disease by shaping protective root-associated microbiomes and metabolites in continuous cropping soils
DOI:10.1007/s11104-022-05532-z [本文引用: 2]
Microbial assembly and association network in watermelon rhizosphere after soil fumigation for Fusarium wilt control
DOI:10.1016/j.agee.2021.107336 URL [本文引用: 5]
Soil texture, sampling depth and root hairs shape the structure of ACC deaminase bacterial community composition in maize rhizosphere
DOI:10.3389/fmicb.2021.616828
URL
[本文引用: 1]
Preservation of the phytostimulatory functions of plant growth-promoting bacteria relies on the adaptation of their community to the rhizosphere environment. Here, an amplicon sequencing approach was implemented to specifically target microorganisms with 1-aminocyclopropane-1-carboxylate deaminase activity, carrying the acdS gene. We stated the hypothesis that the relative phylogenetic distribution of acdS carrying microorganisms is affected by the presence or absence of root hairs, soil type, and depth. To this end, a standardized soil column experiment was conducted with maize wild type and root hair defective rth3 mutant in the substrates loam and sand, and harvest was implemented from three depths. Most acdS sequences (99%) were affiliated to Actinobacteria and Proteobacteria, and the strongest influence on the relative abundances of sequences were exerted by the substrate. Variovorax, Acidovorax, and Ralstonia sequences dominated in loam, whereas Streptomyces and Agromyces were more abundant in sand. Soil depth caused strong variations in acdS sequence distribution, with differential levels in the relative abundances of acdS sequences affiliated to Tetrasphaera, Amycolatopsis, and Streptomyces in loam, but Burkholderia, Paraburkholderia, and Variovorax in sand. Maize genotype influenced the distribution of acdS sequences mainly in loam and only in the uppermost depth. Variovorax acdS sequences were more abundant in WT, but Streptomyces, Microbacterium, and Modestobacter in rth3 rhizosphere. Substrate and soil depth were strong and plant genotype a further significant single and interacting drivers of acdS carrying microbial community composition in the rhizosphere of maize. This suggests that maize rhizosphere acdS carrying bacterial community establishes according to the environmental constraints, and that root hairs possess a minor but significant impact on acdS carrying bacterial populations.
Soil protists: A fertile frontier in soil biology research
DOI:10.1093/femsre/fuy006
PMID:29447350
[本文引用: 1]
Protists include all eukaryotes except plants, fungi and animals. They are an essential, yet often forgotten, component of the soil microbiome. Method developments have now furthered our understanding of the real taxonomic and functional diversity of soil protists. They occupy key roles in microbial foodwebs as consumers of bacteria, fungi and other small eukaryotes. As parasites of plants, animals and even of larger protists, they regulate populations and shape communities. Pathogenic forms play a major role in public health issues as human parasites, or act as agricultural pests. Predatory soil protists release nutrients enhancing plant growth. Soil protists are of key importance for our understanding of eukaryotic evolution and microbial biogeography. Soil protists are also useful in applied research as bioindicators of soil quality, as models in ecotoxicology and as potential biofertilizers and biocontrol agents. In this review, we provide an overview of the enormous morphological, taxonomical and functional diversity of soil protists, and discuss current challenges and opportunities in soil protistology. Research in soil biology would clearly benefit from incorporating more protistology alongside the study of bacteria, fungi and animals.
Phyllosphere microbiota: Community dynamics and its interaction with plant hosts
DOI:10.1111/jipb.13060
[本文引用: 2]
Plants are colonized by various microorganisms in natural environments. While many studies have demonstrated key roles of the rhizosphere microbiota in regulating biological processes such as nutrient acquisition and resistance against abiotic and biotic challenges, less is known about the role of the phyllosphere microbiota and how it is established and maintained. This review provides an update on current understanding of phyllosphere community assembly and the mechanisms by which plants and microbes establish the phyllosphere microbiota for plant health.
Competition for iron drives phytopathogen control by natural rhizosphere microbiomes
DOI:10.1038/s41564-020-0719-8
PMID:32393858
[本文引用: 1]
Plant pathogenic bacteria cause high crop and economic losses to human societies. Infections by such pathogens are challenging to control as they often arise through complex interactions between plants, pathogens and the plant microbiome. Experimental studies of this natural ecosystem at the microbiome-wide scale are rare, and consequently we have a poor understanding of how the taxonomic and functional microbiome composition and the resulting ecological interactions affect pathogen growth and disease outbreak. Here, we combine DNA-based soil microbiome analysis with in vitro and in planta bioassays to show that competition for iron via secreted siderophore molecules is a good predictor of microbe-pathogen interactions and plant protection. We examined the ability of 2,150 individual bacterial members of 80 rhizosphere microbiomes, covering all major phylogenetic lineages, to suppress the bacterium Ralstonia solanacearum, a global phytopathogen capable of infecting various crops. We found that secreted siderophores altered microbiome-pathogen interactions from complete pathogen suppression to strong facilitation. Rhizosphere microbiome members with growth-inhibitory siderophores could often suppress the pathogen in vitro as well as in natural and greenhouse soils, and protect tomato plants from infection. Conversely, rhizosphere microbiome members with growth-promotive siderophores were often inferior in competition and facilitated plant infection by the pathogen. Because siderophores are a chemically diverse group of molecules, with each siderophore type relying on a compatible receptor for iron uptake, our results suggest that pathogen-suppressive microbiome members produce siderophores that the pathogen cannot use. Our study establishes a causal mechanistic link between microbiome-level competition for iron and plant protection and opens promising avenues to use siderophore-mediated interactions as a tool for microbiome engineering and pathogen control.
Trophic interactions between predatory protists and pathogen-suppressive bacteria impact plant health
DOI:10.1038/s41396-022-01244-5 [本文引用: 2]
Field study reveals core plant microbiota and relative importance of their drivers
DOI:10.1111/1462-2920.14031
PMID:29266641
[本文引用: 2]
Harnessing plant microbiota can assist in sustainably increasing primary productivity to meet growing global demands for food and biofuel. However, development of rational microbiome-based approaches for improving crop yield and productivity is currently hindered by a lack of understanding of the major biotic and abiotic factors shaping the crop microbiome under relevant field conditions. We examined bacterial and fungal communities associated with both aerial (leaves, stalks) and belowground (roots, soil) compartments of four commercial sugarcane varieties (Saccharum spp.) grown in several growing regions in Australia. We identified drivers of the sugarcane microbiome under field conditions and evaluated whether the plants shared a core microbiome. Sugarcane-associated microbial assemblages were primarily determined by plant compartment, followed by growing region, crop age, variety and Yellow Canopy Syndrome (YCS). We detected a core set of microbiota and identified members of the core microbiome that were influenced by YCS incidence. Our study revealed key hub microorganisms in the core microbiome networks of sugarcane leaves, stalks, roots and rhizosphere soil despite location and time-associated shifts in the community assemblages. Elucidating their functional roles and identification of the keystone core microbiota that sustain plant health could provide a technological breakthrough for a sustainable increase in crop productivity.© 2017 Society for Applied Microbiology and John Wiley & Sons Ltd.
Origin and evolution of the plant immune system
DOI:10.1111/nph.2019.222.issue-1 URL [本文引用: 1]
13C pulse-labeling assessment of the community structure of active fungi in the rhizosphere of a genetically starch-modified potato (Solanum tuberosum) cultivar and its parental isoline
DOI:10.1111/j.1469-8137.2012.04089.x
PMID:22413848
[本文引用: 1]
• The aim of this study was to gain understanding of the carbon flow from the roots of a genetically modified (GM) amylopectin-accumulating potato (Solanum tuberosum) cultivar and its parental isoline to the soil fungal community using stable isotope probing (SIP). • The microbes receiving (13)C from the plant were assessed through RNA/phospholipid fatty acid analysis with stable isotope probing (PLFA-SIP) at three time-points (1, 5 and 12 d after the start of labeling). The communities of Ascomycota, Basidiomycota and Glomeromycota were analysed separately with RT-qPCR and terminal restriction fragment length polymorphism (T-RFLP). • Ascomycetes and glomeromycetes received carbon from the plant as early as 1 and 5 d after labeling, while basidiomycetes were slower in accumulating the labeled carbon. The rate of carbon allocation in the GM variety differed from that in its parental variety, thereby affecting soil fungal communities. • We conclude that both saprotrophic and mycorrhizal fungi rapidly metabolize organic substrates flowing from the root into the rhizosphere, that there are large differences in utilization of root-derived compounds at a lower phylogenetic level within investigated fungal phyla, and that active communities in the rhizosphere differ between the GM plant and its parental cultivar through effects of differential carbon flow from the plant.© 2012 The Authors. New Phytologist © 2012 New Phytologist Trust.
Root-secreted coumarins and the microbiota interact to improve iron nutrition in Arabidopsis
Properties of bacterial endophytes and their proposed role in plant growth
DOI:10.1016/j.tim.2008.07.008
PMID:18789693
[本文引用: 1]
Bacterial endophytes live inside plants for at least part of their life cycle. Studies of the interaction of endophytes with their host plants and their function within their hosts are important to address the ecological relevance of endophytes. The modulation of ethylene levels in plants by bacterially produced 1-aminocyclopropane-1-carboxylate deaminase is a key trait that enables interference with the physiology of the host plant. Endophytes with this capacity might profit from association with the plant, because colonization is enhanced. In turn, host plants benefit by stress reduction and increased root growth. This mechanism leads to the concept of 'competent' endophytes, defined as endophytes that are equipped with genes important for maintenance of plant-endophyte associations. The ecological role of these endophytes and their relevance for plant growth are discussed here.
The role of flavonoids in root-rhizosphere signalling: Opportunities and challenges for improving plant-microbe interactions
DOI:10.1093/jxb/err430
PMID:22213816
[本文引用: 1]
The flavonoid pathway produces a diverse array of plant compounds with functions in UV protection, as antioxidants, pigments, auxin transport regulators, defence compounds against pathogens and during signalling in symbiosis. This review highlights some of the known function of flavonoids in the rhizosphere, in particular for the interaction of roots with microorganisms. Depending on their structure, flavonoids have been shown to stimulate or inhibit rhizobial nod gene expression, cause chemoattraction of rhizobia towards the root, inhibit root pathogens, stimulate mycorrhizal spore germination and hyphal branching, mediate allelopathic interactions between plants, affect quorum sensing, and chelate soil nutrients. Therefore, the manipulation of the flavonoid pathway to synthesize specifically certain products has been suggested as an avenue to improve root-rhizosphere interactions. Possible strategies to alter flavonoid exudation to the rhizosphere are discussed. Possible challenges in that endeavour include limited knowledge of the mechanisms that regulate flavonoid transport and exudation, unforeseen effects of altering parts of the flavonoid synthesis pathway on fluxes elsewhere in the pathway, spatial heterogeneity of flavonoid exudation along the root, as well as alteration of flavonoid products by microorganisms in the soil. In addition, the overlapping functions of many flavonoids as stimulators of functions in one organism and inhibitors of another suggests caution in attempts to manipulate flavonoid rhizosphere signals.
Microbial interactions within the plant holobiont
DOI:10.1186/s40168-018-0445-0
PMID:29587885
[本文引用: 1]
Since the colonization of land by ancestral plant lineages 450 million years ago, plants and their associated microbes have been interacting with each other, forming an assemblage of species that is often referred to as a "holobiont." Selective pressure acting on holobiont components has likely shaped plant-associated microbial communities and selected for host-adapted microorganisms that impact plant fitness. However, the high microbial densities detected on plant tissues, together with the fast generation time of microbes and their more ancient origin compared to their host, suggest that microbe-microbe interactions are also important selective forces sculpting complex microbial assemblages in the phyllosphere, rhizosphere, and plant endosphere compartments. Reductionist approaches conducted under laboratory conditions have been critical to decipher the strategies used by specific microbes to cooperate and compete within or outside plant tissues. Nonetheless, our understanding of these microbial interactions in shaping more complex plant-associated microbial communities, along with their relevance for host health in a more natural context, remains sparse. Using examples obtained from reductionist and community-level approaches, we discuss the fundamental role of microbe-microbe interactions (prokaryotes and micro-eukaryotes) for microbial community structure and plant health. We provide a conceptual framework illustrating that interactions among microbiota members are critical for the establishment and the maintenance of host-microbial homeostasis.
Ecological assembly processes of the bacterial and fungal microbiota of wild and domesticated wheat species
DOI:10.1094/PBIOMES-01-20-0001-SC
URL
[本文引用: 1]
Domestication has led to substantial changes in plant physiology. How this anthropogenic intervention has contributed in altering the wheat microbiota is not well understood. Here, we investigated the role of ecological selection, drift, and dispersal in shaping the bacterial and fungal communities associated with domesticated wheat Triticum aestivum and two wild relatives, T. boeoticum and T. urartu. Our study shows that the bacterial and fungal microbiota of wild and domesticated wheat species follow distinct community assembly patterns. Further, we revealed a more prominent role of neutral processes in the assembly of the microbiota of domesticated wheat and propose that domestication has relaxed selective processes in the assembly of the wheat microbiota.
Network mapping of root-microbe interactions in Arabidopsis thaliana
DOI:10.1038/s41522-021-00241-4
[本文引用: 1]
Understanding how plants interact with their colonizing microbiota to determine plant phenotypes is a fundamental question in modern plant science. Existing approaches for genome-wide association studies (GWAS) are often focused on the association analysis between host genes and the abundance of individual microbes, failing to characterize the genetic bases of microbial interactions that are thought to be important for microbiota structure, organization, and function. Here, we implement a behavioral model to quantify various patterns of microbe-microbe interactions, i.e., mutualism, antagonism, aggression, and altruism, and map host genes that modulate microbial networks constituted by these interaction types. We reanalyze a root-microbiome data involving 179 accessions of Arabidopsis thaliana and find that the four networks differ structurally in the pattern of bacterial-fungal interactions and microbiome complexity. We identify several fungus and bacterial hubs that play a central role in mediating microbial community assembly surrounding A. thaliana root systems. We detect 1142 significant host genetic variants throughout the plant genome and then implement Bayesian networks (BN) to reconstruct epistatic networks involving all significant SNPs, of which 91 are identified as hub QTLs. Results from gene annotation analysis suggest that most of the hub QTLs detected are in proximity to candidate genes, executing a variety of biological functions in plant growth and development, resilience against pathogens, root development, and abiotic stress resistance. This study provides a new gateway to understand how genetic variation in host plants influences microbial communities and our results could help improve crops by harnessing soil microbes.
Rust fungi and global change
Rust fungi are important components of ecological communities and in ecosystem function. Their unique life strategies as biotrophic pathogens with complicated life cycles could make them vulnerable to global environmental change. While there are gaps in our knowledge, especially in natural plant–rust systems, this review of the exposure of rust fungi to global change parameters revealed that some host–rust relationships would decline under predicted environmental change scenarios, whereas others would either remain unchanged or become more prevalent. Notably, some graminicolous rusts are negatively affected by higher temperatures and increased concentrations of atmospheric CO2. An increase of atmospheric O3 appears to favour rust diseases on trees but not those on grasses. Combined effects of CO2 and O3 are intermediary. The most important global drivers for the geographical and host plant range expansion and prevalence of rusts, however, are global plant trade, host plant genetic homogenization and the regular occurrence of conducive environmental conditions, especially the availability of moisture. However, while rusts thrive in high-humidity environments, they can also survive in desert habitats, and as a group their environmental tolerance is large, with no conclusive change in their overall prevalence predictable to date.
Root endophyte Colletotrichum tofieldiae confers plant fitness benefits that are phosphate status dependent
DOI:10.1016/j.cell.2016.02.028
PMID:26997485
[本文引用: 1]
A staggering diversity of endophytic fungi associate with healthy plants in nature, but it is usually unclear whether these represent stochastic encounters or provide host fitness benefits. Although most characterized species of the fungal genus Colletotrichum are destructive pathogens, we show here that C. tofieldiae (Ct) is an endemic endophyte in natural Arabidopsis thaliana populations in central Spain. Colonization by Ct initiates in roots but can also spread systemically into shoots. Ct transfers the macronutrient phosphorus to shoots, promotes plant growth, and increases fertility only under phosphorus-deficient conditions, a nutrient status that might have facilitated the transition from pathogenic to beneficial lifestyles. The host's phosphate starvation response (PSR) system controls Ct root colonization and is needed for plant growth promotion (PGP). PGP also requires PEN2-dependent indole glucosinolate metabolism, a component of innate immune responses, indicating a functional link between innate immunity and the PSR system during beneficial interactions with Ct.Copyright © 2016 The Authors. Published by Elsevier Inc. All rights reserved.
Root exudate metabolites drive plant-soil feedbacks on growth and defense by shaping the rhizosphere microbiota
DOI:10.1038/s41467-018-05122-7
PMID:30013066
[本文引用: 1]
By changing soil properties, plants can modify their growth environment. Although the soil microbiota is known to play a key role in the resulting plant-soil feedbacks, the proximal mechanisms underlying this phenomenon remain unknown. We found that benzoxazinoids, a class of defensive secondary metabolites that are released by roots of cereals such as wheat and maize, alter root-associated fungal and bacterial communities, decrease plant growth, increase jasmonate signaling and plant defenses, and suppress herbivore performance in the next plant generation. Complementation experiments demonstrate that the benzoxazinoid breakdown product 6-methoxy-benzoxazolin-2-one (MBOA), which accumulates in the soil during the conditioning phase, is both sufficient and necessary to trigger the observed phenotypic changes. Sterilization, fungal and bacterial profiling and complementation experiments reveal that MBOA acts indirectly by altering root-associated microbiota. Our results reveal a mechanism by which plants determine the composition of rhizosphere microbiota, plant performance and plant-herbivore interactions of the next generation.
Insect herbivory reshapes a native leaf microbiome
Bacterial community assemblages in the rhizosphere soil, root endosphere and cyst of soybean cyst nematode-suppressive soil challenged with nematodes
Characterization of the maize gene sugary1, a determinant of starch composition in kernels
DOI:10.1105/tpc.7.4.417
PMID:7773016
[本文引用: 1]
In maize kernels, mutations in the gene sugary1 (su1) result in (1) increased sucrose concentration; (2) decreased concentration of amylopectin, the branched component of starch; and (3) accumulation of the highly branched glucopolysaccharide phytoglycogen. To investigate further the mechanisms of storage carbohydrate synthesis in maize, part of the su1 gene locus and a cDNA copy of the su1 transcript were characterized. Five new su1 mutations were isolated in a Mutator background, and the mutant allele su1-R4582::Mu1 was isolated by transposon tagging. The identity of the cloned element as the su1 gene locus was confirmed by the cosegregation of restriction fragment length polymorphisms in the same or nearby genomic intervals with three additional, independent su1 mutations. Pedigree analysis was also used to confirm the identity of su1. A 2.8-kb mRNA that is homologous to the cloned gene was detected in maize kernels, and a 2.7-kb cDNA clone was isolated based on hybridization to the genomic DNA. Specific portions of the cDNA hybridized with multiple segments of the maize genome, suggesting that su1 is part of a multigene family. The cDNA sequence specified a polypeptide of at least 742 amino acids, which is highly similar in amino acid sequence to bacterial enzymes that hydrolyze alpha-(1-->6) glucosyl linkages of starch. Therefore, debranching of glucopolysaccharides is seemingly part of the normal process of starch biosynthesis, and the final degree of branch linkages in starch most likely arises from the combined actions of branching and debranching enzymes.
Tree species traits affect which natural enemies drive the Janzen-Connell effect in a temperate forest
DOI:10.1038/s41467-019-14140-y
PMID:31941904
[本文引用: 1]
A prominent tree species coexistence mechanism suggests host-specific natural enemies inhibit seedling recruitment at high conspecific density (negative conspecific density dependence). Natural-enemy-mediated conspecific density dependence affects numerous tree populations, but its strength varies substantially among species. Understanding how conspecific density dependence varies with species' traits and influences the dynamics of whole communities remains a challenge. Using a three-year manipulative community-scale experiment in a temperate forest, we show that plant-associated fungi, and to a lesser extent insect herbivores, reduce seedling recruitment and survival at high adult conspecific density. Plant-associated fungi are primarily responsible for reducing seedling recruitment near conspecific adults in ectomycorrhizal and shade-tolerant species. Insects, in contrast, primarily inhibit seedling recruitment of shade-intolerant species near conspecific adults. Our results suggest that natural enemies drive conspecific density dependence in this temperate forest and that which natural enemies are responsible depends on the mycorrhizal association and shade tolerance of tree species.
Different behaviour of methanogenic Archaea and Thaumarchaeota in rice field microcosms
DOI:10.1111/1574-6941.12188
PMID:23909555
[本文引用: 1]
Archaea in rice fields play an important role in carbon and nitrogen cycling. They comprise methane-producing Euryarchaeota as well as ammonia-oxidizing Thaumarchaeota, but their community structures and population dynamics have not yet been studied in the same system. Different soil compartments (surface, bulk, rhizospheric soil) and ages of roots (young and old roots) at two N fertilization levels and at three time points (the panicle initiation, heading and maturity periods) of the season were assayed by determining the abundance (using qPCR) and composition (using T-RFLP and cloning/sequencing) of archaeal genes (mcrA, amoA, 16S rRNA gene). The community of total Archaea in soil and root samples mainly consisted of the methanogens and the Thaumarchaeota and their abundance increased over the season. Methanogens proliferated everywhere, but Thaumarchaeota proliferated only on the roots and in response to nitrogen fertilization. The community structures of Archaea, methanogens and Thaumarchaeota were different in soil and root samples indicating niche differentiation. While Methanobacteriales were generally present, Methanosarcinaceae and Methanocellales were the dominant methanogens in soil and root samples, respectively. The results emphasize the specific colonization of roots by two ecophysiologically different groups of archaea which may belong to the core root biome.© 2013 Federation of European Microbiological Societies. Published by John Wiley & Sons Ltd. All rights reserved.
Relationships between phyllosphere bacterial communities and plant functional traits in a neotropical forest
Domestication of Oryza species eco-evolutionarily shapes bacterial and fungal communities in rice seed
DOI:10.1186/s40168-020-00805-0
URL
[本文引用: 1]
Plant-associated microbiomes, which are shaped by host and environmental factors, support their hosts by providing nutrients and attenuating abiotic and biotic stresses. Although host genetic factors involved in plant growth and immunity are known to shape compositions of microbial communities, the effects of host evolution on microbial communities are not well understood.
Metaproteogenomic analysis of microbial communities in the phyllosphere and rhizosphere of rice
DOI:10.1038/ismej.2011.192 [本文引用: 1]
Rhizosphere microbiome structure alters to enable wilt resistance in tomato
DOI:10.1038/nbt.4232 URL [本文引用: 3]
Host species identity, site and time drive temperate tree phyllosphere bacterial community structure
DOI:10.1186/s40168-016-0174-1
PMID:27316353
[本文引用: 1]
Background: The increasing awareness of the role of phyllosphere microbial communities in plant health calls for a greater understanding of their structure and dynamics in natural ecosystems. Since most knowledge of tree phyllosphere bacterial communities has been gathered in tropical forests, our goal was to characterize the community structure and assembly dynamics of phyllosphere epiphytic bacterial communities in temperate forests in Quebec, Canada. We targeted five dominant tree species: Acer saccharum, Acer rubrum, Betula papyrifera, Abies balsamea, and Picea glauca. We collected 180 samples of phyllosphere communities on these species at four natural forest sites, three times during the growing season. Results: Host functional traits (i.e., wood density, leaf nitrogen content) and climate variables (summer mean temperature and precipitation) were strongly correlated with community structure. We highlight three key findings: (1) temperate tree species share a "core microbiome"; (2) significant evolutionary associations exist between groups of bacteria and host species; and (3) a greater part of the variation in phyllosphere bacterial community assembly is explained by host species identity (27 %) and species-site interaction (14 %), than by site (11 %) or time (1 %). Conclusions: We demonstrated that host species identity is a stronger driver of temperate tree phyllosphere bacterial communities than site or time. Our results suggest avenues for future studies on the influence of host functional traits on phyllosphere community functional biogeography across terrestrial biomes.
Communication in the phytobiome
DOI:S0092-8674(17)30476-2
PMID:28475891
[本文引用: 2]
The phytobiome is composed of plants, their environment, and diverse interacting microscopic and macroscopic organisms, which together influence plant health and productivity. These organisms form complex networks that are established and regulated through nutrient cycling, competition, antagonism, and chemical communication mediated by a diverse array of signaling molecules. Integration of knowledge of signaling mechanisms with that of phytobiome members and their networks will lead to a new understanding of the fate and significance of these signals at the ecosystem level. Such an understanding could lead to new biological, chemical, and breeding strategies to improve crop health and productivity.Copyright © 2017 Elsevier Inc. All rights reserved.
Disruption of Firmicutes and Actinobacteria abundance in tomato rhizosphere causes the incidence of bacterial wilt disease
DOI:10.1038/s41396-020-00785-x
[本文引用: 2]
Enrichment of protective microbiota in the rhizosphere facilitates disease suppression. However, how the disruption of protective rhizobacteria affects disease suppression is largely unknown. Here, we analyzed the rhizosphere microbial community of a healthy and diseased tomato plant grown <30-cm apart in a greenhouse at three different locations in South Korea. The abundance of Gram-positive Actinobacteria and Firmicutes phyla was lower in diseased rhizosphere soil (DRS) than in healthy rhizosphere soil (HRS) without changes in the causative Ralstonia solanacearum population. Artificial disruption of Gram-positive bacteria in HRS using 500-μg/mL vancomycin increased bacterial wilt occurrence in tomato. To identify HRS-specific and plant-protective Gram-positive bacteria species, Brevibacterium frigoritolerans HRS1, Bacillus niacini HRS2, Solibacillus silvestris HRS3, and Bacillus luciferensis HRS4 were selected from among 326 heat-stable culturable bacteria isolates. These four strains did not directly antagonize R. solanacearum but activated plant immunity. A synthetic community comprising these four strains displayed greater immune activation against R. solanacearum and extended plant protection by 4 more days in comparison with each individual strain. Overall, our results demonstrate for the first time that dysbiosis of the protective Gram-positive bacterial community in DRS promotes the incidence of disease.
Genomic features of bacterial adaptation to plants
DOI:10.1038/s41588-017-0012-9
[本文引用: 2]
Plants intimately associate with diverse bacteria. Plant-associated bacteria have ostensibly evolved genes that enable them to adapt to plant environments. However, the identities of such genes are mostly unknown, and their functions are poorly characterized. We sequenced 484 genomes of bacterial isolates from roots of Brassicaceae, poplar, and maize. We then compared 3,837 bacterial genomes to identify thousands of plant-associated gene clusters. Genomes of plant-associated bacteria encode more carbohydrate metabolism functions and fewer mobile elements than related non-plant-associated genomes do. We experimentally validated candidates from two sets of plant-associated genes: one involved in plant colonization, and the other serving in microbe-microbe competition between plant-associated bacteria. We also identified 64 plant-associated protein domains that potentially mimic plant domains; some are shared with plant-associated fungi and oomycetes. This work expands the genome-based understanding of plant-microbe interactions and provides potential leads for efficient and sustainable agriculture through microbiome engineering.
The phyllosphere microbiome shifts toward combating melanose pathogen
DOI:10.1186/s40168-022-01234-x
URL
[本文引用: 1]
Plants can recruit beneficial microbes to enhance their ability to defend against pathogens. However, in contrast to the intensively studied roles of the rhizosphere microbiome in suppressing plant pathogens, the collective community-level change and effect of the phyllosphere microbiome in response to pathogen invasion remains largely elusive.
Characterization of diazotrophic root endophytes in Chinese silvergrass (Miscanthus sinensis)
DOI:10.1186/s40168-022-01379-9
[本文引用: 1]
Phytoremediation is a potentially cost-effective way to remediate highly contaminated mine tailing sites. However, nutrient limitations, especially the deficiency of nitrogen (N), can hinder the growth of plants and impair the phytoremediation of mine tailings. Nevertheless, pioneer plants can successfully colonize mine tailings and exhibit potential for tailing phytoremediation. Diazotrophs, especially diazotrophic endophytes, can promote the growth of their host plants. This was tested in a mine-tailing habitat by a combination of field sampling, DNA-stable isotope probing (SIP) analysis, and pot experiments.
A simplified synthetic community rescues Astragalus mongholicus from root rot disease by activating plant-induced systemic resistance
DOI:10.1186/s40168-021-01169-9
URL
[本文引用: 1]
Plant health and growth are negatively affected by pathogen invasion; however, plants can dynamically modulate their rhizosphere microbiome and adapt to such biotic stresses. Although plant-recruited protective microbes can be assembled into synthetic communities for application in the control of plant disease, rhizosphere microbial communities commonly contain some taxa at low abundance. The roles of low-abundance microbes in synthetic communities remain unclear; it is also unclear whether all the microbes enriched by plants can enhance host adaptation to the environment. Here, we assembled a synthetic community with a disease resistance function based on differential analysis of root-associated bacterial community composition. We further simplified the synthetic community and investigated the roles of low-abundance bacteria in the control of Astragalus mongholicus root rot disease by a simple synthetic community.
Microbiology of the phyllosphere
DOI:10.1128/AEM.69.4.1875-1883.2003 PMID:12676659 [本文引用: 1]
Linking the phyllosphere microbiome to plant health
DOI:S1360-1385(20)30199-0
PMID:32576433
[本文引用: 1]
The phyllosphere harbors diverse microbial communities that influence ecosystem functioning. Emerging evidence suggests that plants impaired in genetic networks harbor an altered microbiome and develop dysbiosis in the phyllosphere, which pinpoints plant genetics as a key driver of the phyllosphere microbiome assembly and links the phyllosphere microbiome to plant health.Copyright © 2020 Elsevier Ltd. All rights reserved.
Evidence for the plant recruitment of beneficial microbes to suppress soil-borne pathogens
DOI:10.1111/nph.v229.5 URL [本文引用: 1]
The effect of foliar fungal pathogens on plant species coexistence: Progress and challenge
DOI:10.17520/biods.2022525 URL [本文引用: 1]
叶片病原真菌对植物物种共存的影响: 进展与挑战
DOI:10.17520/biods.2022525
[本文引用: 1]
群落内物种如何共存是群落生态学研究中最具争议的核心问题之一。根据当代物种共存理论框架, 维持物种共存的机制可以分为稳定化和均等化机制。尽管植物叶片病原真菌在自然界中大量存在, 但是目前尚不完全清楚叶片病原真菌如何通过稳定化和均等化机制影响物种共存。本文首先介绍了叶片病原真菌驱动同种负密度制约(稳定化机制)和“生长-防御”权衡(均等化机制)促进物种共存的证据, 并阐述了在群落水平抑制叶片病原真菌后物种丰富度的变化。随后, 本文归纳了该领域研究中的主要挑战: 叶片病原真菌在驱动物种共存过程中相较于环境因子的可能重要性更低、部分叶片病原真菌较弱的宿主专一性无法起到维持物种共存的作用, 以及控制叶片病原真菌过程中各种方法均有一定局限性等问题。最后, 本文论述了该研究领域未来的主要方向: 不同土壤养分/气候变化条件下叶片病原真菌如何影响物种共存、叶片病原真菌与其他高营养层次生物类群的交互作用及其对物种共存的贡献、基于系统发育推断叶片病原真菌对植物群落构建的影响、将叶片病原真菌生活史类型纳入病原真菌影响植物物种共存的相关研究中。
Leaf-residing Methylobacterium species fix nitrogen and promote biomass and seed production in Jatropha curcas
DOI:10.1186/s13068-015-0404-y
PMID:26697111
[本文引用: 2]
Background: Jatropha curcas L. (Jatropha) is a potential biodiesel crop that can be cultivated on marginal land because of its strong tolerance to drought and low soil nutrient content. However, seed yield remains low. To enhance the commercial viability and green index of Jatropha biofuel, a systemic and coordinated approach must be adopted to improve seed oil and biomass productivity. Here, we present our investigations on the Jatropha-associated nitrogen-fixing bacteria with an aim to understand and exploit the unique biology of this plant from the perspective of plant-microbe interactions. Results: An analysis of 1017 endophytic bacterial isolates derived from different parts of Jatropha revealed that diazotrophs were abundant and diversely distributed into five classes belonging to alpha, beta, gamma-Proteobacteria, Actinobacteria and Firmicutes. Methylobacterium species accounted for 69.1 % of endophytic bacterial isolates in leaves and surprisingly, 30.2 % which were able to fix nitrogen that inhabit in leaves. Among the Methylobacterium isolates, strain L2-4 was characterized in detail. Phylogenetically, strain L2-4 is closely related to M. radiotolerans and showed strong molybdenum-iron dependent acetylene reduction (AR) activity in vitro and in planta. Foliar spray of L2-4 led to successful colonization on both leaf surface and in internal tissues of systemic leaves and significantly improved plant height, leaf number, chlorophyll content and stem volume. Importantly, seed production was improved by 222.2 and 96.3 % in plants potted in sterilized and non-sterilized soil, respectively. Seed yield increase was associated with an increase in female-male flower ratio. Conclusion: The ability of Methylobacterium to fix nitrogen and colonize leaf tissues serves as an important trait for Jatropha. This bacteria-plant interaction may significantly contribute to Jatropha's tolerance to low soil nutrient content. Strain L2-4 opens a new possibility to improve plant's nitrogen supply from the leaves and may be exploited to significantly improve the productivity and Green Index of Jatropha biofuel.
The fungal root endophyte Serendipita vermifera displays inter-Kingdom synergistic beneficial effects with the microbiota in Arabidopsis thaliana and barley
DOI:10.1038/s41396-021-01138-y
[本文引用: 1]
Plant root-associated bacteria can confer protection against pathogen infection. By contrast, the beneficial effects of root endophytic fungi and their synergistic interactions with bacteria remain poorly defined. We demonstrate that the combined action of a fungal root endophyte from a widespread taxon with core bacterial microbiota members provides synergistic protection against an aggressive soil-borne pathogen inArabidopsis thalianaand barley. We additionally reveal early inter-kingdom growth promotion benefits which are host and microbiota composition dependent. Using RNA-sequencing, we show that these beneficial activities are not associated with extensive host transcriptional reprogramming but rather with the modulation of expression of microbial effectors and carbohydrate-active enzymes.
Influence of resistance breeding in common bean on rhizosphere microbiome composition and function
DOI:10.1038/ismej.2017.158 URL [本文引用: 3]
The rhizosphere microbiome: Significance of plant beneficial, plant pathogenic, and human pathogenic microorganisms
DOI:10.1111/1574-6976.12028
PMID:23790204
[本文引用: 1]
Microbial communities play a pivotal role in the functioning of plants by influencing their physiology and development. While many members of the rhizosphere microbiome are beneficial to plant growth, also plant pathogenic microorganisms colonize the rhizosphere striving to break through the protective microbial shield and to overcome the innate plant defense mechanisms in order to cause disease. A third group of microorganisms that can be found in the rhizosphere are the true and opportunistic human pathogenic bacteria, which can be carried on or in plant tissue and may cause disease when introduced into debilitated humans. Although the importance of the rhizosphere microbiome for plant growth has been widely recognized, for the vast majority of rhizosphere microorganisms no knowledge exists. To enhance plant growth and health, it is essential to know which microorganism is present in the rhizosphere microbiome and what they are doing. Here, we review the main functions of rhizosphere microorganisms and how they impact on health and disease. We discuss the mechanisms involved in the multitrophic interactions and chemical dialogues that occur in the rhizosphere. Finally, we highlight several strategies to redirect or reshape the rhizosphere microbiome in favor of microorganisms that are beneficial to plant growth and health. © 2013 Federation of European Microbiological Societies. Published by John Wiley & Sons Ltd. All rights reserved.
Deciphering the rhizosphere microbiome for disease-suppressive bacteria
DOI:10.1126/science.1203980
PMID:21551032
[本文引用: 1]
Disease-suppressive soils are exceptional ecosystems in which crop plants suffer less from specific soil-borne pathogens than expected owing to the activities of other soil microorganisms. For most disease-suppressive soils, the microbes and mechanisms involved in pathogen control are unknown. By coupling PhyloChip-based metagenomics of the rhizosphere microbiome with culture-dependent functional analyses, we identified key bacterial taxa and genes involved in suppression of a fungal root pathogen. More than 33,000 bacterial and archaeal species were detected, with Proteobacteria, Firmicutes, and Actinobacteria consistently associated with disease suppression. Members of the γ-Proteobacteria were shown to have disease-suppressive activity governed by nonribosomal peptide synthetases. Our data indicate that upon attack by a fungal root pathogen, plants can exploit microbial consortia from soil for protection against infections.
Archaea are interactive components of complex microbiomes
DOI:S0966-842X(17)30174-9
PMID:28826642
[本文引用: 1]
Recent findings have shaken our picture of the biology of the archaea and revealed novel traits beyond archaeal extremophily and supposed 'primitiveness'. The archaea constitute a considerable fraction of the Earth's ecosystems, and their potential to shape their surroundings by a profound interaction with their biotic and abiotic environment has been recognized. Moreover, archaea have been identified as a substantial component, or even as keystone species, in complex microbiomes - in the environment or accompanying a holobiont. Species of the Euryarchaeota (methanogens, halophiles) and Thaumarchaeota, in particular, have the capacity to coexist in plant, animal, and human microbiomes, where syntrophy allows them to thrive under energy-deficiency stress. Due to methodological limitations, the archaeome remains mysterious, and many questions with respect to potential pathogenicity, function, and structural interactions with their host and other microorganisms remain.Copyright © 2017 Elsevier Ltd. All rights reserved.
The impact of bacteriophages on phyllosphere bacterial abundance and composition
DOI:10.1111/mec.14542
PMID:29457297
[本文引用: 1]
Interactions between bacteria and bacteriophage viruses (phages) are known to influence pathogen growth and virulence, microbial diversity and even biogeochemical cycling. Lytic phages in particular infect and lyse their host cells, and can therefore have significant effects on cell densities as well as competitive dynamics within microbial communities. Despite the known impacts of lytic phages on the ecology and evolution of bacteria in free-living communities, little is known about the role of lytic phages in host-associated microbiomes. We set out to characterize the impact of phages in the tomato phyllosphere, that is the bacteria associated with above-ground plant tissues, by transferring microbial communities from field-grown tomato plants to juvenile plants grown under mostly sterile conditions in either the presence or absence of their associated phage community. In three separate experiments, we found that the presence of phages affects overall bacterial abundance during colonization of new host plants. Furthermore, bacterial community analysis using 16S rRNA amplicon sequencing shows that phages significantly alter the relative abundance of dominant community members and can influence both within- and among-host diversity. These results underscore the importance of lytic phages in host-associated microbiomes and are relevant to microbiome transplantation approaches, as they suggest transferring nonbacterial components of the microbiome among hosts is likely to have a strong impact on growth of both the resident and colonizing microbiota.© 2018 John Wiley & Sons Ltd.
Successive passaging of a plant-associated microbiome reveals robust habitat and host genotype-dependent selection
The plant microbiota: Systems-level insights and perspectives
Plant genotype-specific archaeal and bacterial endophytes but similar Bacillus antagonists colonize Mediterranean olive trees
DOI:10.3389/fmicb.2015.00138
PMID:25784898
[本文引用: 1]
Endophytes have an intimate and often symbiotic interaction with their hosts. Less is known about the composition and function of endophytes in trees. In order to evaluate our hypothesis that plant genotype and origin have a strong impact on both, endophytes of leaves from 10 Olea europaea L. cultivars from the Mediterranean basin growing at a single agricultural site in Spain and from nine wild olive trees located in natural habitats in Greece, Cyprus, and on Madeira Island were studied. The composition of the bacterial endophytic communities as revealed by 16S rRNA gene amplicon sequencing and the subsequent PCoA analysis showed a strong correlation to the plant genotypes. The bacterial distribution patterns were congruent with the plant origins in "Eastern" and "Western" areas of the Mediterranean basin. Subsequently, the endophytic microbiome of wild olives was shown to be closely related to those of cultivated olives of the corresponding geographic origins. The olive leaf endosphere harbored mostly Proteobacteria, followed by Firmicutes, Actinobacteria, and Bacteroidetes.The detection of a high portion of archaeal taxa belonging to the phyla Thaumarchaeota, Crenarchaeota, and Euryarchaeota in the amplicon libraries was an unexpected discovery, which was confirmed by quantitative real-time PCR revealing an archaeal portion of up to 35.8%. Although the function of these Archaea for their host plant remains speculative, this finding suggests a significant relevance of archaeal endophytes for plant microbe interactions. In addition, the antagonistic potential of culturable endophytes was determined; all isolates with antagonistic activity against the olive-pathogenic fungus Verticillium dahliae Kleb. belong to Bacillus amyloliquefaciens. In contrast to the specific global structural diversity, BOX-fingerprints of the antagonistic Bacillus isolates were highly similar and independent of the olive genotype from which they were isolated.
Patterns and processes of microbial community assembly
DOI:10.1128/MMBR.00051-12
PMID:24006468
[本文引用: 1]
Recent research has expanded our understanding of microbial community assembly. However, the field of community ecology is inaccessible to many microbial ecologists because of inconsistent and often confusing terminology as well as unnecessarily polarizing debates. Thus, we review recent literature on microbial community assembly, using the framework of Vellend (Q. Rev. Biol. 85:183-206, 2010) in an effort to synthesize and unify these contributions. We begin by discussing patterns in microbial biogeography and then describe four basic processes (diversification, dispersal, selection, and drift) that contribute to community assembly. We also discuss different combinations of these processes and where and when they may be most important for shaping microbial communities. The spatial and temporal scales of microbial community assembly are also discussed in relation to assembly processes. Throughout this review paper, we highlight differences between microbes and macroorganisms and generate hypotheses describing how these differences may be important for community assembly. We end by discussing the implications of microbial assembly processes for ecosystem function and biodiversity.
Simplified and representative bacterial community of maize roots
Diversity and heritability of the maize rhizosphere microbiome under field conditions
Linking rhizosphere microbiome composition of wild and domesticated Phaseolus vulgaris to genotypic and root phenotypic traits
DOI:10.1038/ismej.2017.85 URL [本文引用: 1]
The wild side of plant microbiomes
DOI:10.1186/s40168-018-0519-z
PMID:30115122
[本文引用: 1]
<空>
Going back to the roots: The microbial ecology of the rhizosphere
DOI:10.1038/nrmicro3109
PMID:24056930
[本文引用: 4]
The rhizosphere is the interface between plant roots and soil where interactions among a myriad of microorganisms and invertebrates affect biogeochemical cycling, plant growth and tolerance to biotic and abiotic stress. The rhizosphere is intriguingly complex and dynamic, and understanding its ecology and evolution is key to enhancing plant productivity and ecosystem functioning. Novel insights into key factors and evolutionary processes shaping the rhizosphere microbiome will greatly benefit from integrating reductionist and systems-based approaches in both agricultural and natural ecosystems. Here, we discuss recent developments in rhizosphere research in relation to assessing the contribution of the micro- and macroflora to sustainable agriculture, nature conservation, the development of bio-energy crops and the mitigation of climate change.
Induced systemic resistance by beneficial microbes
DOI:10.1146/annurev-phyto-082712-102340
PMID:24906124
[本文引用: 1]
Beneficial microbes in the microbiome of plant roots improve plant health. Induced systemic resistance (ISR) emerged as an important mechanism by which selected plant growth-promoting bacteria and fungi in the rhizosphere prime the whole plant body for enhanced defense against a broad range of pathogens and insect herbivores. A wide variety of root-associated mutualists, including Pseudomonas, Bacillus, Trichoderma, and mycorrhiza species sensitize the plant immune system for enhanced defense without directly activating costly defenses. This review focuses on molecular processes at the interface between plant roots and ISR-eliciting mutualists, and on the progress in our understanding of ISR signaling and systemic defense priming. The central role of the root-specific transcription factor MYB72 in the onset of ISR and the role of phytohormones and defense regulatory proteins in the expression of ISR in aboveground plant parts are highlighted. Finally, the ecological function of ISR-inducing microbes in the root microbiome is discussed.
Agriculture and the disruption of plant-microbial symbiosis
DOI:S0169-5347(20)30008-2
PMID:32294424
[本文引用: 1]
Domestication has transformed hundreds of wild plant species into productive cultivars for human utility. However, cultivation practices and intense artificial selection for yield may entail a hidden cost: the disruption of interactions between plants and beneficial microbiota. Here, we synthesize theory predicting that evolutionary trade-offs, genetic costs, and relaxed selection disrupt plant-microbial symbiosis under domestication, and review the wealth of new data interrogating these predictions in crops. We describe the agronomic practices, ecological scenarios, and genomic attributes that can result in the disruption of symbiosis, and highlight new work probing its molecular basis. To improve agricultural output and sustainability, research should develop breeding methods to optimize symbiotic outcomes in crop species.Published by Elsevier Ltd.
The ‘neglected’ soil virome—Potential role and impact
DOI:S0966-842X(17)30277-9
PMID:29306554
[本文引用: 1]
Bacteriophages are among the most abundant and diverse biological units in the biosphere. They have contributed to our understanding of the central dogma of biology and have been instrumental in the evolutionary success of bacterial pathogens. In contrast to our current understanding of marine viral communities, the soil virome and its function in terrestrial ecosystems has remained relatively understudied. Here, we examine, in a comparative fashion, the knowledge gathered from studies performed in soil versus marine settings. We address the information with respect to the abundance, diversity, ecological significance, and effects of, in particular, bacteriophages on their host's evolutionary trajectories. We also identify the main challenges that soil virology faces and the studies that are required to accompany the current developments in marine settings.Copyright © 2017 Elsevier Ltd. All rights reserved.
Microbially mediated plant salt tolerance and microbiome-based solutions for saline agriculture
DOI:S0734-9750(16)30106-9
PMID:27587331
[本文引用: 2]
Soil salinization adversely affects plant growth and has become one of the major limiting factors for crop productivity worldwide. The conventional approach, breeding salt-tolerant plant cultivars, has often failed to efficiently alleviate the situation. In contrast, the use of a diverse array of microorganisms harbored by plants has attracted increasing attention because of the remarkable beneficial effects of microorganisms on plants. Multiple advanced '-omics' technologies have enabled us to gain insights into the structure and function of plant-associated microbes. In this review, we first focus on microbe-mediated plant salt tolerance, in particular on the physiological and molecular mechanisms underlying root-microbe symbiosis. Unfortunately, when introducing such microbes as single strains to soils, they are often ineffective in improving plant growth and stress tolerance, largely due to competition with native soil microbial communities and limited colonization efficiency. Rapid progress in rhizosphere microbiome research has revived the belief that plants may benefit more from association with interacting, diverse microbial communities (microbiome) than from individual members in a community. Understanding how a microbiome assembles in the continuous compartments (endosphere, rhizoplane, and rhizosphere) will assist in predicting a subset of core or minimal microbiome and thus facilitate synthetic re-construction of microbial communities and their functional complementarity and synergistic effects. These developments will open a new avenue for capitalizing on the cultivable microbiome to strengthen plant salt tolerance and thus to refine agricultural practices and production under saline conditions.Copyright © 2016 Elsevier Inc. All rights reserved.
Rewilding plant microbiomes
DOI:10.1126/science.abn6350
PMID:36356130
[本文引用: 1]
Microbiota of crop ancestors may offer a way to enhance sustainable food production.
New insights into the structure and function of phyllosphere microbiota through high-throughput molecular approaches
DOI:10.1111/1574-6968.12225
PMID:23895412
[本文引用: 1]
The phyllosphere is an ecologically and economically important ecosystem that hosts a large and diverse microbial community. Phyllosphere microbiota play a critical role in protecting plants from diseases as well as promoting their growth by various mechanisms. There are serious gaps in our understanding of how and why microbiota composition varies across spatial and temporal scales, the ecology of leaf surface colonizers and their interactions with their host, and the genetic adaptations that enable phyllosphere survival of microorganisms. These gaps are due in large part to past technical limitations, as earlier studies were restricted to the study of culturable bacteria only and used low-throughput molecular techniques to describe community structure and function. The availability of high-throughput and cost-effective molecular technologies is changing the field of phyllosphere microbiology, enabling researchers to begin to address the dynamics and composition of the phyllosphere microbiota across a large number of samples with high, in-depth coverage. Here, we discuss and connect the most recent studies that have used next-generation molecular techniques such as metagenomics, proteogenomics, genome sequencing, and transcriptomics to gain new insights into the structure and function of phyllosphere microbiota and highlight important challenges for future research. © 2013 Federation of European Microbiological Societies. Published by John Wiley & Sons Ltd. All rights reserved.
Root-associated microorganisms reprogram plant life history along the growth-stress resistance tradeoff
DOI:10.1038/s41396-019-0501-1 [本文引用: 2]
Phyllosphere microbiology: At the interface between microbial individuals and the plant host
DOI:10.1111/nph.15054
PMID:29504646
[本文引用: 1]
Contents Summary 1327 I. Introduction 1327 II. Individuality and the relevance of scales for the investigation of bacteria 1328 III. Bacterial aggregation and community patterning at the single-cell resolution 1329 IV. What are the effects on the plant host? 1330 V. Future directions and current questions 1331 Acknowledgements 1332 ORCID 1332 References 1332 SUMMARY: Leaf surfaces are home to diverse bacterial communities. Within these communities, every individual cell perceives its unique environment and responds accordingly. In this insight article, the perspective of the bacterial individual is assumed in an attempt to describe how the spatially heterogeneous leaf surface determines the fate of bacteria. To investigate behaviour at scales relevant to bacteria, single-cell approaches are essential. Single-cell studies provide important lessons about how current 'omics' approaches fail to give an accurate picture of the behaviour of bacterial populations in heterogeneous environments. Upcoming techniques will soon allow us to combine the power of single-cell and omics approaches.© 2018 The Authors. New Phytologist © 2018 New Phytologist Trust.
Complexities of microbial life on leaf surfaces
DOI:10.1128/microbe.9.448.1 URL [本文引用: 1]
Acquisition of phosphorus and nitrogen in the rhizosphere and plant growth promotion by microorganisms
DOI:10.1007/s11104-009-9895-2 URL [本文引用: 2]
Root-secreted malic acid recruits beneficial soil bacteria
DOI:10.1104/pp.108.127613
PMID:18820082
[本文引用: 1]
Beneficial soil bacteria confer immunity against a wide range of foliar diseases by activating plant defenses, thereby reducing a plant's susceptibility to pathogen attack. Although bacterial signals have been identified that activate these plant defenses, plant metabolites that elicit rhizobacterial responses have not been demonstrated. Here, we provide biochemical evidence that the tricarboxylic acid cycle intermediate L-malic acid (MA) secreted from roots of Arabidopsis (Arabidopsis thaliana) selectively signals and recruits the beneficial rhizobacterium Bacillus subtilis FB17 in a dose-dependent manner. Root secretions of L-MA are induced by the foliar pathogen Pseudomonas syringae pv tomato (Pst DC3000) and elevated levels of L-MA promote binding and biofilm formation of FB17 on Arabidopsis roots. The demonstration that roots selectively secrete L-MA and effectively signal beneficial rhizobacteria establishes a regulatory role of root metabolites in recruitment of beneficial microbes, as well as underscores the breadth and sophistication of plant-microbial interactions.
Understanding the holobiont: The interdependence of plants and their microbiome
DOI:S1369-5274(17)30042-5
PMID:28732267
[本文引用: 1]
The holobiont is composed by the plant and its microbiome. In a similar way to ecological systems of higher organisms, the holobiont shows interdependent and complex dynamics [1,2]. While plants originate from seeds, the microbiome has a multitude of sources. The assemblage of these communities depends on the interaction between the emerging seedling and its surrounding environment, with soil being the main source. These microbial communities are controlled by the plant through different strategies, such as the specific profile of root exudates and its immune system. Despite this control, the microbiome is still able to adapt and thrive. The molecular knowledge behind these interactions and microbial '-omic' technologies are developing to the point of enabling holobiont engineering. For a long time microorganisms were in the background of plant biology but new multidisciplinary approaches have led to an appreciation of the importance of the holobiont, where plants and microbes are interdependent.Crown Copyright © 2017. Published by Elsevier Ltd. All rights reserved.
Prolonged drought imparts lasting compositional changes to the rice root microbiome
DOI:10.1038/s41477-021-00967-1
PMID:34294907
[本文引用: 2]
Microbial symbioses can mitigate drought stress in crops but harnessing these beneficial interactions will require an in-depth understanding of root microbiome responses to drought cycles. Here, by detailed temporal characterization of root-associated microbiomes of rice plants during drought stress and recovery, we find that endosphere communities remained compositionally altered after rewatering, with prolonged droughts leading to decreased resilience. Several endospheric Actinobacteria were significantly enriched during drought and for weeks after rewatering. Notably, the most abundant endosphere taxon during this period was a Streptomyces, and a corresponding isolate promoted root growth. Additionally, drought stress disrupted the temporal dynamics of late-colonizing microorganisms, permanently altering the normal successional trends of root microbiota. These findings reveal that severe drought results in enduring impacts on rice root microbiomes, including enrichment of taxonomic groups that could shape the recovery response of the host, and have implications relevant to drought protection strategies using root microbiota.© 2021. The Author(s), under exclusive licence to Springer Nature Limited.
Host genotype is an important determinant of the cereal phyllosphere mycobiome
DOI:10.1111/nph.13418
PMID:25898906
[本文引用: 1]
The phyllosphere mycobiome in cereals is an important determinant of crop health. However, an understanding of the factors shaping this community is lacking. Fungal diversity in leaves from a range of cultivars of winter wheat (Triticum aestivum), winter and spring barley (Hordeum vulgare) and a smaller number of samples from oat (Avena sativa), rye (Secale cereale) and triticale (Triticum × Secale) was studied using next-generation sequencing. The effects of host genotype, fungicide treatment and location on fungal communities were explored. In total, 635 251 fungal internal transcribed spacer (ITS) reads were obtained from 210 leaf samples. Visual disease assessments and relative read abundance of Zymoseptoria tritici and Ramularia collo-cygni were strongly positively related. Crop genotype at the species level explained 43% of the variance in the total dataset, followed by fungicide treatment (13%) and location (4%). Indicator species, including plant pathogens, responding to factors such as crop species, location and treatment were identified. Host genotype at both the species and cultivar level is important in shaping phyllosphere fungal communities, whereas fungicide treatment and location have minor effects. We found many host-specific fungal pathogens, but also a large diversity of fungi that were relatively insensitive to host genetic background, indicating that host-specific pathogens live in a 'sea' of nonspecific fungi.© 2015 The Authors. New Phytologist © 2015 New Phytologist Trust.
Protists are an integral part of the Arabidopsis thaliana microbiome
DOI:10.1111/emi.2018.20.issue-1 URL [本文引用: 1]
The inconspicuous gatekeeper: Endophytic Serendipita vermifera acts as extended plant protection barrier in the rhizosphere
DOI:10.1111/nph.v224.2 URL [本文引用: 1]
Feed your friends: Do plant exudates shape the root microbiome?
DOI:S1360-1385(17)30199-1
PMID:29050989
[本文引用: 3]
Plant health in natural environments depends on interactions with complex and dynamic communities comprising macro- and microorganisms. While many studies have provided insights into the composition of rhizosphere microbiomes (rhizobiomes), little is known about whether plants shape their rhizobiomes. Here, we discuss physiological factors of plants that may govern plant-microbe interactions, focusing on root physiology and the role of root exudates. Given that only a few plant transport proteins are known to be involved in root metabolite export, we suggest novel families putatively involved in this process. Finally, building off of the features discussed in this review, and in analogy to well-known symbioses, we elaborate on a possible sequence of events governing rhizobiome assembly.Copyright © 2017 Elsevier Ltd. All rights reserved.
Synthetic bacterial community derived from a desert rhizosphere confers salt stress resilience to tomato in the presence of a soil microbiome
DOI:10.1038/s41396-022-01238-3
[本文引用: 1]
The root bacterial microbiome is important for the general health of the plant. Additionally, it can enhance tolerance to abiotic stresses, exemplified by plant species found in extreme ecological niches like deserts. These complex microbe-plant interactions can be simplified by constructing synthetic bacterial communities or SynComs from the root microbiome. Furthermore, SynComs can be applied as biocontrol agents to protect crops against abiotic stresses such as high salinity. However, there is little knowledge on the design of a SynCom that offers a consistent protection against salt stress for plants growing in a natural and, therefore, non-sterile soil which is more realistic to an agricultural setting. Here we show that a SynCom of five bacterial strains, originating from the root of the desert plant Indigofera argentea, protected tomato plants growing in a non-sterile substrate against a high salt stress. This phenotype correlated with the differential expression of salt stress related genes and ion accumulation in tomato. Quantification of the SynCom strains indicated a low penetrance into the natural soil used as the non-sterile substrate. Our results demonstrate how a desert microbiome could be engineered into a simplified SynCom that protected tomato plants growing in a natural soil against an abiotic stress.
Ecological patterns of seed microbiome diversity, transmission, and assembly
DOI:S1369-5274(16)30157-6
PMID:28437661
[本文引用: 1]
Seeds are involved in the transmission of microorganisms from one plant generation to another and consequently act as the initial inoculum for the plant microbiota. The purpose of this mini-review is to provide an overview of current knowledge on the diversity, structure and role of the seed microbiota. The relative importance of the mode of transmission (vertical vs horizontal) of the microbial entities composing the seed microbiota as well as the potential connections existing between seed and other plant habitats such as the anthosphere and the spermosphere is discussed. Finally the governing processes (niche vs neutral) involved in the assembly and the dynamics of the seed microbiota are examined.Copyright © 2017. Published by Elsevier Ltd.
Fertilization alters protistan consumers and parasites in crop-associated microbiomes
DOI:10.1111/1462-2920.15385
PMID:33400366
[本文引用: 1]
Crop plants carry an enormous diversity of microbiota that provide massive benefits to hosts. Protists, as the main microbial consumers and a pivotal driver of biogeochemical cycling processes, remain largely understudied in the plant microbiome. Here, we characterized the diversity and composition of protists in sorghum leaf phyllosphere, and rhizosphere and bulk soils, collected from an 8-year field experiment with multiple fertilization regimes. Phyllosphere was an important habitat for protists, dominated by Rhizaria, Alveolata and Amoebozoa. Rhizosphere and bulk soils had a significantly higher diversity of protists than the phyllosphere, and the protistan community structure significantly differed among the three plant-soil compartments. Fertilization significantly altered specific functional groups of protistan consumers and parasites. Variation partitioning models revealed that soil properties, bacteria and fungi predicted a significant proportion of the variation in the protistan communities. Changes in protists may in turn significantly alter the compositions of bacterial and fungal communities from the top-down control in food webs. Altogether, we provide novel evidence that fertilization significantly affects the functional groups of protistan consumers and parasites in crop-associated microbiomes, which have implications for the potential changes in their ecological functions under intensive agricultural managements.© 2021 Society for Applied Microbiology and John Wiley & Sons Ltd.
Root traits explain rhizosphere fungal community composition among temperate grassland plant species
DOI:10.1111/nph.v229.3 URL [本文引用: 2]
Root microbiota assembly and adaptive differentiation among European Arabidopsis populations
A cosmopolitan fungal pathogen of dicots adopts an endophytic lifestyle on cereal crops and protects them from major fungal diseases
DOI:10.1038/s41396-020-00744-6
[本文引用: 2]
Fungal pathogens are seriously threatening food security and natural ecosystems; efficient and environmentally friendly control methods are essential to help safeguard such resources for increasing human populations on a global scale. Here, we find that Sclerotinia sclerotiorum, a widespread pathogen of dicotyledons, can grow endophytically in wheat, rice, barley, maize, and oat, providing protection against Fusarium head blight, stripe rust, and rice blast. Protection is also provided by disabled S. sclerotiorum strains harboring a hypovirulence virus. The disabled strain DT-8 promoted wheat yields by 4–18% in the field and consistently reduced Fusarium disease by 40–60% across multiple field trials. We term the host-dependent trophism of S. sclerotiorum, destructively pathogenic or mutualistically endophytic, as schizotrophism. As a biotroph, S. sclerotiorum modified the expression of wheat genes involved in disease resistance and photosynthesis and increased the level of IAA. Our study shows that a broad-spectrum pathogen of one group of plants may be employed as a biocontrol agent in a different group of plants where they can be utilized as beneficial microorganisms while avoiding the risk of in-field release of pathogens. Our study also raises provocative questions about the potential role of schizotrophic endophytes in natural ecosystems.
Core microbiomes for sustainable agroecosystems
DOI:10.1038/s41477-018-0139-4
PMID:29725101
[本文引用: 1]
In an era of ecosystem degradation and climate change, maximizing microbial functions in agroecosystems has become a prerequisite for the future of global agriculture. However, managing species-rich communities of plant-associated microbiomes remains a major challenge. Here, we propose interdisciplinary research strategies to optimize microbiome functions in agroecosystems. Informatics now allows us to identify members and characteristics of 'core microbiomes', which may be deployed to organize otherwise uncontrollable dynamics of resident microbiomes. Integration of microfluidics, robotics and machine learning provides novel ways to capitalize on core microbiomes for increasing resource-efficiency and stress-resistance of agroecosystems.
Plant-nematode interactions assisted by microbes in the rhizosphere
DOI:10.21775/cimb.030.075
PMID:30070652
[本文引用: 1]
Plant health is strongly influenced by the interactions between parasites/pathogens and beneficial microorganisms. In this chapter we will summarize the up-to date knowledge on soil suppressiveness as a biological tool against phytonematodes and explore the nature of monoculture versus crop rotation in this regard. Since nematodes are successfully antagonized by different microbiological agents, we highlighted this phenomenon with respect to the most important antagonists, and a nature of these interactions. The focus is on the hyperparasitic microbes of phytonematodes such as Pasteuria sp. and egg parasites. Furthermore, we comprised the studies on the defence system expressions in plants triggered by nematode-associated microbes. The attachment of bacteria and fungi to phytonematodes and putative effects of the attachment on the induced systemic resistance in plants are discussed. Finally, our chapter is rounded up with the importance of incorporating the knowledge on plant-nematode-microbe interactions in the integrated pest management.
Plant-microbiome interactions under a changing world: Responses, consequences and perspectives
DOI:10.1111/nph.18016
PMID:35118660
[本文引用: 2]
Climate change is increasing global temperatures and the frequency and severity of droughts in many regions. These anthropogenic stresses pose a significant threat to plant performance and crop production. The plant-associated microbiome modulates the impacts of biotic and abiotic stresses on plant fitness. However, climate change-induced alteration in composition and activities of plant microbiomes can affect host functions. Here, we highlight recent advancements in our understanding of the impact of climate change (warming and drought) on plant-microbiome interactions and on their ecological functions from genome to ecosystem scales. We identify knowledge gaps, propose new concepts and make recommendations for future research directions. It is proposed that in the short term (years to decades), the adaptation of plants to climate change is mainly driven by the plant microbiome, whereas in the long term (century to millennia), the adaptation of plants will be driven equally by eco-evolutionary interactions between the plant microbiome and its host. A better understanding of the response of the plant and its microbiome interactions to climate change and the ways in which microbiomes can mitigate the negative impacts will better inform predictions of climate change impacts on primary productivity and aid in developing management and policy tools to improve the resilience of plant systems.© 2022 The Authors New Phytologist © 2022 New Phytologist Foundation.
Plant-microbiome interactions: From community assembly to plant health
DOI:10.1038/s41579-020-0412-1 [本文引用: 7]
Continuum of root-fungal symbioses for plant nutrition
Mycorrhizal ecology and evolution: The past, the present, and the future
DOI:10.1111/nph.13288
PMID:25639293
[本文引用: 1]
Almost all land plants form symbiotic associations with mycorrhizal fungi. These below-ground fungi play a key role in terrestrial ecosystems as they regulate nutrient and carbon cycles, and influence soil structure and ecosystem multifunctionality. Up to 80% of plant N and P is provided by mycorrhizal fungi and many plant species depend on these symbionts for growth and survival. Estimates suggest that there are c. 50 000 fungal species that form mycorrhizal associations with c. 250 000 plant species. The development of high-throughput molecular tools has helped us to better understand the biology, evolution, and biodiversity of mycorrhizal associations. Nuclear genome assemblies and gene annotations of 33 mycorrhizal fungal species are now available providing fascinating opportunities to deepen our understanding of the mycorrhizal lifestyle, the metabolic capabilities of these plant symbionts, the molecular dialogue between symbionts, and evolutionary adaptations across a range of mycorrhizal associations. Large-scale molecular surveys have provided novel insights into the diversity, spatial and temporal dynamics of mycorrhizal fungal communities. At the ecological level, network theory makes it possible to analyze interactions between plant-fungal partners as complex underground multi-species networks. Our analysis suggests that nestedness, modularity and specificity of mycorrhizal networks vary and depend on mycorrhizal type. Mechanistic models explaining partner choice, resource exchange, and coevolution in mycorrhizal associations have been developed and are being tested. This review ends with major frontiers for further research.© 2015 The Authors. New Phytologist © 2015 New Phytologist Trust.
Modelling sugar diffusion across plant leaf cuticles: the effect of free water on substrate availability to phyllosphere bacteria
DOI:10.1111/j.1462-2920.2010.02382.x
PMID:21091864
[本文引用: 1]
We present a continuous model for the diffusion of sugars across intact plant leaf cuticles. It is based on the flow of sugars from a source, representing the leaf apoplast, to a sink, in the shape of a hemispherical drop of water on the outside of the cuticle. Flow is a function of the difference between sugar concentrations C(Source) and C(Sink), permeability P of the cuticle, volume V(Sink) of the water drop, as well as its contact angle α with the cuticle surface. Using a bacterial bioreporter for fructose, and a two-compartment experimental set-up consisting of isolated cuticles of walnut (Juglans regia) carrying water droplets while floating on solutions with increasing concentrations of fructose, we determined a value of 1 × 10⁻⁶ m h⁻¹ for P. Using this value, we explored different scenarios for the leaching of sugars across plant leaf cuticles to reveal in quantitative terms how diffusion takes longer when V(Sink) increases, P decreases or α increases. Bacterial growth was modelled as a function of changes in P, α and V(Sink) and was consistent with observations or suggestions from the literature in relation to the availability of free water on leaves. These results are discussed in the light of bacteria as ecosystem engineers, i.e. with the ability to modify the plant leaf surface environment in favour of their own survival, e.g. by increasing cuticle leakage or leaf wetness. Our model represents a first step towards a more comprehensive model which will enhance our quantitative understanding of the factors that play a role in nutrient availability to bacterial colonizers of the phyllosphere, or plant leaf surface.© 2010 Society for Applied Microbiology and Blackwell Publishing Ltd.
Nitrogen fixation in a landrace of maize is supported by a mucilage-associated diazotrophic microbiota
DOI:10.1371/journal.pbio.2006352 URL [本文引用: 1]
Active root-inhabiting microbes identified by rapid incorporation of plant-derived carbon into RNA
The importance of the microbiome of the plant holobiont
DOI:10.1111/nph.13312
PMID:25655016
[本文引用: 2]
Plants can no longer be considered as standalone entities and a more holistic perception is needed. Indeed, plants harbor a wide diversity of microorganisms both inside and outside their tissues, in the endosphere and ectosphere, respectively. These microorganisms, which mostly belong to Bacteria and Fungi, are involved in major functions such as plant nutrition and plant resistance to biotic and abiotic stresses. Hence, the microbiota impact plant growth and survival, two key components of fitness. Plant fitness is therefore a consequence of the plant per se and its microbiota, which collectively form a holobiont. Complementary to the reductionist perception of evolutionary pressures acting on plant or symbiotic compartments, the plant holobiont concept requires a novel perception of evolution. The interlinkages between the plant holobiont components are explored here in the light of current ecological and evolutionary theories. Microbiome complexity and the rules of microbiotic community assemblage are not yet fully understood. It is suggested that the plant can modulate its microbiota to dynamically adjust to its environment. To better understand the level of plant dependence on the microbiotic components, the core microbiota need to be determined at different hierarchical scales of ecology while pan-microbiome analyses would improve characterization of the functions displayed.© 2015 The Authors New Phytologist © 2015 New Phytologist Trust.
Conceptual synthesis in community ecology
DOI:10.1086/652373 URL [本文引用: 1]
Plant-derived coumarins shape the composition of an Arabidopsis synthetic root microbiome
Microbial life in the phyllosphere
DOI:10.1038/nrmicro2910
PMID:23154261
[本文引用: 1]
Our knowledge of the microbiology of the phyllosphere, or the aerial parts of plants, has historically lagged behind our knowledge of the microbiology of the rhizosphere, or the below-ground habitat of plants, particularly with respect to fundamental questions such as which microorganisms are present and what they do there. In recent years, however, this has begun to change. Cultivation-independent studies have revealed that a few bacterial phyla predominate in the phyllosphere of different plants and that plant factors are involved in shaping these phyllosphere communities, which feature specific adaptations and exhibit multipartite relationships both with host plants and among community members. Insights into the underlying structural principles of indigenous microbial phyllosphere populations will help us to develop a deeper understanding of the phyllosphere microbiota and will have applications in the promotion of plant growth and plant protection.
Host genotype and age shape the leaf and root microbiomes of a wild perennial plant
DOI:10.1038/ncomms12151
PMID:27402057
[本文引用: 2]
Bacteria living on and in leaves and roots influence many aspects of plant health, so the extent of a plant's genetic control over its microbiota is of great interest to crop breeders and evolutionary biologists. Laboratory-based studies, because they poorly simulate true environmental heterogeneity, may misestimate or totally miss the influence of certain host genes on the microbiome. Here we report a large-scale field experiment to disentangle the effects of genotype, environment, age and year of harvest on bacterial communities associated with leaves and roots of Boechera stricta (Brassicaceae), a perennial wild mustard. Host genetic control of the microbiome is evident in leaves but not roots, and varies substantially among sites. Microbiome composition also shifts as plants age. Furthermore, a large proportion of leaf bacterial groups are shared with roots, suggesting inoculation from soil. Our results demonstrate how genotype-by-environment interactions contribute to the complexity of microbiome assembly in natural environments.
Large-scale replicated field study of maize rhizosphere identifies heritable microbes
Soil Protozoa: Research methods and roles in the biocontrol of soil-borne diseases
土壤原生动物——研究方法及其在土传病害防控中的作用
Microbial populations responsible for specific soil suppressiveness to plant pathogens
Agricultural soils suppressive to soilborne plant pathogens occur worldwide, and for several of these soils the biological basis of suppressiveness has been described. Two classical types of suppressiveness are known. General suppression owes its activity to the total microbial biomass in soil and is not transferable between soils. Specific suppression owes its activity to the effects of individual or select groups of microorganisms and is transferable. The microbial basis of specific suppression to four diseases, Fusarium wilts, potato scab, apple replant disease, and take-all, is discussed. One of the best-described examples occurs in take-all decline soils. In Washington State, take-all decline results from the buildup of fluorescent Pseudomonas spp. that produce the antifungal metabolite 2,4-diacetylphloroglucinol. Producers of this metabolite may have a broader role in disease-suppressive soils worldwide. By coupling molecular technologies with traditional approaches used in plant pathology and microbiology, it is possible to dissect the microbial composition and complex interactions in suppressive soils.
Rare taxa maintain the stability of crop mycobiomes and ecosystem functions
DOI:10.1111/emi.v23.4 URL [本文引用: 5]
Plant developmental stage drives the differentiation in ecological role of the maize microbiome
DOI:10.1186/s40168-021-01118-6
URL
[本文引用: 5]
Plants live with diverse microbial communities which profoundly affect multiple facets of host performance, but if and how host development impacts the assembly, functions and microbial interactions of crop microbiomes are poorly understood. Here we examined both bacterial and fungal communities across soils, epiphytic and endophytic niches of leaf and root, and plastic leaf of fake plant (representing environment-originating microbes) at three developmental stages of maize at two contrasting sites, and further explored the potential function of phylloplane microbiomes based on metagenomics.
Host selection shapes crop microbiome assembly and network complexity
Rhizosphere protists are key determinants of plant health
DOI:10.1186/s40168-020-00799-9
PMID:32127034
[本文引用: 1]
Plant health is intimately influenced by the rhizosphere microbiome, a complex assembly of organisms that changes markedly across plant growth. However, most rhizosphere microbiome research has focused on fractions of this microbiome, particularly bacteria and fungi. It remains unknown how other microbial components, especially key microbiome predators-protists-are linked to plant health. Here, we investigated the holistic rhizosphere microbiome including bacteria, microbial eukaryotes (fungi and protists), as well as functional microbial metabolism genes. We investigated these communities and functional genes throughout the growth of tomato plants that either developed disease symptoms or remained healthy under field conditions.We found that pathogen dynamics across plant growth is best predicted by protists. More specifically, communities of microbial-feeding phagotrophic protists differed between later healthy and diseased plants at plant establishment. The relative abundance of these phagotrophs negatively correlated with pathogen abundance across plant growth, suggesting that predator-prey interactions influence pathogen performance. Furthermore, phagotrophic protists likely shifted bacterial functioning by enhancing pathogen-suppressing secondary metabolite genes involved in mitigating pathogen success.We illustrate the importance of protists as top-down controllers of microbiome functioning linked to plant health. We propose that a holistic microbiome perspective, including bacteria and protists, provides the optimal next step in predicting plant performance. Video Abstract.
Genome-resolved metagenomics reveals role of iron metabolism in drought-induced rhizosphere microbiome dynamics
DOI:10.1038/s41467-021-23553-7
PMID:34050180
[本文引用: 1]
Recent studies have demonstrated that drought leads to dramatic, highly conserved shifts in the root microbiome. At present, the molecular mechanisms underlying these responses remain largely uncharacterized. Here we employ genome-resolved metagenomics and comparative genomics to demonstrate that carbohydrate and secondary metabolite transport functionalities are overrepresented within drought-enriched taxa. These data also reveal that bacterial iron transport and metabolism functionality is highly correlated with drought enrichment. Using time-series root RNA-Seq data, we demonstrate that iron homeostasis within the root is impacted by drought stress, and that loss of a plant phytosiderophore iron transporter impacts microbial community composition, leading to significant increases in the drought-enriched lineage, Actinobacteria. Finally, we show that exogenous application of iron disrupts the drought-induced enrichment of Actinobacteria, as well as their improvement in host phenotype during drought stress. Collectively, our findings implicate iron metabolism in the root microbiome's response to drought and may inform efforts to improve plant drought tolerance to increase food security.
Drought delays development of the sorghum root microbiome and enriches for monoderm bacteria
Research progress on the biodiversity and ecological function of soil protists
DOI:10.17520/biods.2022353
[本文引用: 1]
<p id="p00010"><strong>Background & Aims:</strong> Protists are widely distributed in soil and throughout different habitats with high abundance and diversity. They play important roles in nutrient cycling and the energy flow of ecosystems, as well as maintaining soil and plant health. Compared with other microorganisms and fauna in soil, protists have received little attention until recently, and the study on their classification and molecular detection are largely challenged due to their complex taxonomy systems and ecological types.<br><strong>Progresses:</strong> This review systematically summarized and sorted out previous research on soil protists. The research progress on the taxonomic systems of protists, properties of different trophic functional groups, the distribution pattern, and the influencing factors of soil protists were summarized. Then the ecological functions of protistan communities in participating in soil nutrient cycling and maintaining soil health were further highlighted. The main factors that drive the construction of protistan community was clarified, and the prospect and application prospect were further put forward.<br><strong>Prospects:</strong> The future perspectives and research efforts towards taxonomic classification, biodiversity, ecological function, and applications of soil protists need to be explored.</p>
土壤原生生物多样性及其生态功能研究进展
DOI:10.17520/biods.2022353
[本文引用: 1]
原生生物广泛分布在土壤和不同生境中, 其数量庞大、种类繁多, 在生态系统物质循环和能量流动以及维持土壤和植物健康中起着举足轻重的作用。与土壤其他生物类群相比, 原生生物分类体系和生态类型复杂, 分类鉴定困难且分子检测技术不够成熟, 目前对原生生物的认识相对不足。本文对当前原生生物的相关研究进展进行了总结和梳理, 系统阐述了原生生物的分类系统和营养功能群特征、土壤原生生物的多样性分布特征及影响因子, 重点介绍了原生生物群落在参与土壤养分循环、维持土壤和植物健康等中的功能作用, 并对未来的研究方向与应用前景进行了展望。对土壤原生生物的研究有助于我们深入认识土壤生物多样性资源, 并进行保护性地开发和利用, 维护土壤和生态系统健康。
Phyllosphere epiphytic and endophytic fungal community and network structures differ in a tropical mangrove ecosystem
DOI:10.1186/s40168-019-0671-0
PMID:30967154
[本文引用: 1]
Revealing the relationship between plants and fungi is very important in understanding biodiversity maintenance, community stability, and ecosystem functioning. However, differences in the community and network structures of phyllosphere epiphytic and endophytic fungi are currently poorly documented. In this study, we examined epiphytic and endophytic fungal communities associated with the leaves of six mangrove species using Illumina MiSeq sequencing of internal transcribed spacer 2 (ITS2) sequences.A total of 635 operational taxonomic units (OTUs) of endophytic and epiphytic fungi were obtained at a 97% sequence similarity level; they were dominated by Dothideomycetes and Tremellomycetes. Plant identity had a significant effect on the OTU richness of endophytic fungi, but not on epiphytic fungi. The community composition of epiphytic and endophytic fungi was significantly different, and plant identity had a greater effect on endophytic fungi than on epiphytic fungi. Network analysis showed that both epiphytic and endophytic network structures were characterized by significantly highly specialized and modular but lowly connected and anti-nested properties. Furthermore, the endophytic network had higher levels of specialization and modularity but lower connectance and stronger anti-nestedness than the epiphytic network.This study reveals that the phyllosphere epiphytic and endophytic fungal communities differ, and plant identity has a greater effect on the endophytic fungi than on epiphytic fungi. These findings demonstrate the role of host plant identity in driving phyllosphere epiphytic and endophytic community structure.
Phyllosphere Methylobacterium bacteria contain UVA-absorbing compounds
DOI:10.1016/j.jphotobiol.2016.12.019 URL [本文引用: 1]
Plant flavones enrich rhizosphere Oxalobacteraceae to improve maize performance under nitrogen deprivation
DOI:10.1038/s41477-021-00897-y
PMID:33833418
[本文引用: 2]
Beneficial interactions between plant roots and rhizosphere microorganisms are pivotal for plant fitness. Nevertheless, the molecular mechanisms controlling the feedback between root architecture and microbial community structure remain elusive in maize. Here, we demonstrate that transcriptomic gradients along the longitudinal root axis associate with specific shifts in rhizosphere microbial diversity. Moreover, we have established that root-derived flavones predominantly promote the enrichment of bacteria of the taxa Oxalobacteraceae in the rhizosphere, which in turn promote maize growth and nitrogen acquisition. Genetic experiments demonstrate that LRT1-mediated lateral root development coordinates the interactions of the root system with flavone-dependent Oxalobacteraceae under nitrogen deprivation. In summary, these experiments reveal the genetic basis of the reciprocal interactions between root architecture and the composition and diversity of specific microbial taxa in the rhizosphere resulting in improved plant performance. These findings may open new avenues towards the breeding of high-yielding and nutrient-efficient crops by exploiting their interaction with beneficial soil microorganisms.
NRT1.1B is associated with root microbiota composition and nitrogen use in field-grown rice
DOI:10.1038/s41587-019-0104-4
PMID:31036930
[本文引用: 2]
Nitrogen-use efficiency of indica varieties of rice is superior to that of japonica varieties. We apply 16S ribosomal RNA gene profiling to characterize root microbiota of 68 indica and 27 japonica varieties grown in the field. We find that indica and japonica recruit distinct root microbiota. Notably, indica-enriched bacterial taxa are more diverse, and contain more genera with nitrogen metabolism functions, than japonica-enriched taxa. Using genetic approaches, we provide evidence that NRT1.1B, a rice nitrate transporter and sensor, is associated with the recruitment of a large proportion of indica-enriched bacteria. Metagenomic sequencing reveals that the ammonification process is less abundant in the root microbiome of the nrt1.1b mutant. We isolated 1,079 pure bacterial isolates from indica and japonica roots and derived synthetic communities (SynComs). Inoculation of IR24, an indica variety, with an indica-enriched SynCom improved rice growth in organic nitrogen conditions compared with a japonica-enriched SynCom. The links between plant genotype and root microbiota membership established in this study will inform breeding strategies to improve nitrogen use in crops.
A novel archaeal phylum: Thaumarchaeota—A review
一个新的古菌类群——奇古菌门(Thaumarchaeota)
A highly conserved core bacterial microbiota with nitrogen-fixation capacity inhabits the xylem sap in maize plants
DOI:10.1038/s41467-022-31113-w
PMID:35688828
[本文引用: 3]
Microbiomes are important for crop performance. However, a deeper knowledge of crop-associated microbial communities is needed to harness beneficial host-microbe interactions. Here, by assessing the assembly and functions of maize microbiomes across soil types, climate zones, and genotypes, we found that the stem xylem selectively recruits highly conserved microbes dominated by Gammaproteobacteria. We showed that the proportion of bacterial taxa carrying the nitrogenase gene (nifH) was larger in stem xylem than in other organs such as root and leaf endosphere. Of the 25 core bacterial taxa identified in xylem sap, several isolated strains were confirmed to be active nitrogen-fixers or to assist with biological nitrogen fixation. On this basis, we established synthetic communities (SynComs) consisting of two core diazotrophs and two helpers. GFP-tagged strains and N isotopic dilution method demonstrated that these SynComs do thrive and contribute, through biological nitrogen fixation, 11.8% of the total N accumulated in maize stems. These core taxa in xylem sap represent an untapped resource that can be exploited to increase crop productivity.© 2022. The Author(s).
Characterization of the microbial flora and management to induce the disease suppressive soil
抑病型土壤的微生物区系特征及调控
The unseen rhizosphere root-soil-microbe interactions for crop production
DOI:S1369-5274(17)30001-2
PMID:28433932
[本文引用: 1]
The underground root-soil-microbe interactions are extremely complex, but vitally important for aboveground plant growth, health and fitness. The pressure to reduce our reliance on agrochemicals, and sustainable efforts to develop agriculture makes rhizosphere interactions' research a hotspot. Recent advances provide new insights about the signals, pathways, functions and mechanisms of these interactions. In this review, we provide an overview about recent progress in rhizosphere interaction networks in crops. We also discuss a holistic view of the root-soil-rhizomicrobiome interactions achieved through the advances of omics and bioinformatics technologies, and the potential strategies to manage the complex rhizosphere interactions for enhancing crop production.Copyright © 2017 Elsevier Ltd. All rights reserved.
Mycelial network-mediated rhizobial dispersal enhances legume nodulation
DOI:10.1038/s41396-020-0587-5 [本文引用: 2]
Stochastic community assembly: Does it matter in microbial ecology?
Bathyarchaeota: Globally distributed metabolic generalists in anoxic environments
DOI:10.1093/femsre/fuy023
PMID:29790926
[本文引用: 1]
Bathyarchaeota, formerly known as the Miscellaneous Crenarchaeotal Group, is a phylum of global generalists that are widespread in anoxic sediments, which host relatively high abundance archaeal communities. Until now, 25 subgroups have been identified in the Bathyarchaeota. The distinct bathyarchaeotal subgroups diverged to adapt to marine and freshwater environments. Based on the physiological and genomic evidence, acetyl-coenzyme A-centralized heterotrophic pathways of energy conservation have been proposed to function in Bathyarchaeota; these microbes are able to anaerobically utilize (i) detrital proteins, (ii) polymeric carbohydrates, (iii) fatty acids/aromatic compounds, (iv) methane (or short chain alkane) and methylated compounds, and/or (v) potentially other organic matter. Furthermore, bathyarchaeotal members have wide metabolic capabilities, including acetogenesis, methane metabolism, and dissimilatory nitrogen and sulfur reduction, and they also have potential interactions with anaerobic methane-oxidizing archaea, acetoclastic methanogens and heterotrophic bacteria. These results have not only demonstrated multiple and important ecological functions of this archaeal phylum, but also paved the way for a detailed understanding of the evolution and metabolism of archaea as such. This review summarizes the recent findings pertaining to the ecological, physiological and genomic aspects of Bathyarchaeota, highlighting the vital role of this phylum in global carbon cycling.

{kind=link}
{kind=link}
{kind=link}
{kind=link}