



气候变化和人类活动增加了土壤的风蚀、水蚀和盐渍化, 导致全球荒漠化进程加剧。在中亚干旱区, 降水量及降水频率的变化是荒漠化的重要成因之一(陈宝瑞等, 2007)。自20世纪80年代, 受潜在蒸发增加的影响, 中亚干旱区降水格局具有南北反相变化特征, 其荒漠气候地带向北扩延了7.0%, 荒漠面积达到407.68 × 104 km2 (Liu et al, 2022)。中亚干旱区的荒漠化导致区域植被覆盖度减少、物种多样性降低, 以及区域植被的碳储潜力降低(陶冶和张元明, 2013)。土壤微生物对维持干旱荒漠区生物多样性与群落演替非常重要, 降水通过改变荒漠土壤的干湿条件和微生物群落直接影响土壤的生物化学过程, 并进一步影响荒漠植被及其他生物群落的生态适应过程(van Der Heijden et al, 2008; Neilson et al, 2017; Jansson & Hofmockel, 2020)。因此, 明确降水变化对荒漠土壤微生物群落分布特征及群落构建过程的影响, 对预测干旱区生态系统结构和功能的荒漠化响应及反馈机制具有重要意义。
荒漠土壤微生物群落对降水变化的响应受降水差异程度的调控。基于全球尺度114个不同幅度降水减少(减少幅度为5%‒100%不等)控制实验的Meta分析显示, 微生物生物量的响应方式主要由降水减少幅度而不是降水持续时间决定, 在年均降水量(MAP, mean annual precipitation)减少幅度较高(67%‒100%)的站点, 土壤微生物总生物量显著减少26.6%; 相对于干旱草地和灌丛, 森林土壤微生物总生物量减少幅度更大(17.3%), 这可能是由于不同降水减少幅度下的可利用水分存在差异, 从而影响微生物与土壤基质的物理接触。与小幅度降水减少(0‒33%)相比, 大幅度降水减少(67%‒100%)显著影响微生物的底物利用效率, 这在很大程度上限制了土壤微生物的生长。在中、长期降水持续减少的情况下, 微生物种群可能适应水分胁迫及随之增加的温度胁迫, 进一步促使降水减少幅度而不是降水持续时间成为影响微生物总生物量的主要因素(Ren et al, 2018)。在野外样带调查研究表明, 土壤微生物群落在自然降水梯度也受到了降水差异程度的调控。例如, 以色列沙漠(年均降水梯度介于100‒450 mm), 非洲西南部极端干旱的纳米布沙漠(年均降水梯度介于100‒400 mm)的区域小范围年降水减少显著改变了微生物群落组成, 但对微生物群落物种多样性没有显著的影响(Bachar et al, 2010; Naidoo et al, 2021)。然而, 在内蒙古荒漠草原, 年均降水量215‒261 mm是干旱和半干旱草原土壤细菌群落结构显著变化的分界点, 随着年均降水量的增加, 富营养细菌变形菌门和拟杆菌门的相对丰度增加, 而耐干旱细菌放线菌门、绿弯菌门和芽单胞菌门的相对丰度减少(Yao et al, 2017)。这些已有的研究大多基于模拟降水变化的控制试验或基于较大规模野外调查, 而温带荒漠具有典型的高频率小降水的降水模式(王亚婷和唐立松, 2009), 这种小范围历史性降水差异对其土壤细菌多样性及其维持机制, 以及气候变化背景下的荒漠生态系统功能的影响, 我们的认识仍然有限。
不同丰度微生物类群对降水变化表现出不同的响应模式(Delgado-Baquerizo et al, 2018; Jia et al, 2018)。徐鹏等(2022)在古尔班通古特沙漠的定点降水实验表明, 极端降水减少显著改变了低丰度真菌类群的多样性, 而对高丰度真菌类群多样性没有显著影响。在我国北方草原降水样带(年均降水梯度介于159‒516 mm), 相较于高丰度细菌类群, 低丰度细菌群落的β多样性在样带不同降水样点间群落组成差异更大(Yang et al, 2022)。一方面, 这可能是由于低丰度细菌在样带不同降水梯度样点间具有更高的迁移率, 从而使其具有更高的多样性; 另一方面, 低丰度细菌类群比高丰度细菌类群更容易受到环境过滤作用, 对降水变化更敏感(Liu et al, 2015)。Jiao和Lu (2020a)在农田生态系统研究中也发现, 高丰度真菌相较于低丰度真菌, 对环境梯度的生态偏好表现出更宽的响应阈值与强的环境适应性, 低丰度真菌类群则能快速响应环境变化。因此, 分析不同丰度微生物群落对降水变化的响应, 有助于我们进一步认识荒漠微生物群落潜在的生态适应策略和功能。
微生物的群落构建过程是认识不同降水差异下微生物多样性维持机制的重要手段之一, 对预测微生物群落稳定性和生态系统功能之间的联系也非常重要(Hanson et al, 2012; Jiao & Lu, 2020b)。目前, 人们普遍认为微生物群落构建过程由随机性和确定性过程共同决定(Knelman & Nemergut, 2014; Dini-Andreote et al, 2015), 然而, 不同生态系统中确定性过程和随机过程对维持土壤微生物群落多样性的相对重要性还存有比较大的争议(Stegen et al, 2016; Chen et al, 2020; Jiao & Lu, 2020a, b)。大量研究表明在水分受限制的生态系统中, 降水量显著影响群落随机过程和确定性过程之间的平衡(Cao et al, 2016; Xu et al, 2022; Yang et al, 2022), 在较为湿润的森林、湿地和农田等生态系统中, 土壤微生物群落构建多由确定性过程主导, 随机性和确定性过程的平衡主要受到pH、年均温和土壤养分的限制(Jiao & Lu, 2020b; Ni et al, 2021; Pan et al, 2022); 而在荒漠生态系统中, MAP是介导土壤细菌群落多样性维持机制的最主要因素。例如, Yang等(2022)研究发现, MAP介导了高丰度和低丰度细菌类群群落构建过程的动态平衡, 随MAP的减少, 北方荒漠草原随机性过程的相对重要性逐渐增加, 而确定性过程的贡献则减少, 说明水分的可利用性可以调节土壤细菌的群落构建过程。最近, Pan等(2022)发现, 荒漠土壤低于一定的干旱阈值(如1-AI < 0.92), 确定性过程主导高丰度细菌类群的群落构建过程, 随机性过程主导低丰度细菌类群的群落构建。然而, 当土壤处于极端干旱(即1-AI > 0.92)时, 高丰度和低丰度细菌均趋向于更具确定性的构建过程, 可能在较为干旱的土壤中, 随机作用仍然对低丰度微生物类群的物种多样性有益, 其底物限制更多地影响了高丰度微生物类群的多样性。极端干旱土壤促使土壤团聚体空间隔离或异质性持续增加, 低丰度及高丰度微生物类群均受到了强烈的资源限制作用, 导致确定性过程成为高丰度以及低丰度微生物类群多样性维持的主导作用。
古尔班通古特沙漠是我国典型的固定、半固定沙漠, 0‒5 mm的小量级降水频次占总降水频次的89.8% (王亚婷和唐立松, 2009)。沙漠由西向东的降水变化对植被群落产生了一定影响, 整体上, 沙漠西部植物群落多样性与均匀度小于东部(曾勇, 2015①(①曾勇 (2015) 古尔班通古特沙漠植物多样性对降水变化的敏感性研究. 硕士学位论文, 石河子大学, 新疆石河子.); 罗宁等, 2016)。前期本研究团队通过对比干旱3年和干旱10年对荒漠表层土壤真菌群落多样性的影响发现, 极端干旱显著改变了荒漠表层土壤真菌群落组成, 减弱了真菌物种间的相互作用(徐鹏等, 2022)。然而, 对于长期历史性的小范围降水差异对荒漠表层土壤的细菌群落多样性及其群落构建过程产生的影响尚不清楚。为此, 本研究想要进一步探讨: (1)长期小范围的降水差异情况下, 荒漠表层土壤细菌群落的β多样性是否具有显著差异; (2)小范围降水差异是否是影响不同丰度细菌多样性维持的重要因子; (3)不同降水梯度随机过程在荒漠土壤细菌群落构建过程中的变化特征, 以期我们的研究结果能有助于更准确地预测荒漠生态系统如何通过调节地下细菌群落多样性来响应环境变化。
1 研究方法
1.1 研究区概况
古尔班通古特沙漠位于新疆准噶尔盆地中部(44.18°46.33° N, 80.52°-90.00° E, 海拔300‒600 m), 面积4.88 × 104 km2, 是我国最大的固定和半固定沙漠。该沙漠年均降水量50‒250 mm, 年均温19.0‒22.0℃ (1980-2015年) (中国资源环境科学与数据中心,
1.2 样地设置和土壤样品采集
2020年8月, 选取古尔班通古特沙漠西部至东部5个无生物结皮覆盖、年均降水量(MAP)具有一定差异且生境相似的丘间低地作为取样点。彩南西(CNW)、彩南北(CNN)、一站(YZ)、石西南(SXS)和石西东(SXE) 5个样点的MAP分别为181 mm、190 mm、204 mm、221 mm和232 mm (附录1)。在每个样点设置3个30 m × 30 m 的样方, 样方间距 > 100 m, 样方内随机设置2个1 m × 1 m的小样方, 使用环刀五点梅花法采集小样方0-10 cm土层土壤, 混合成1个样品。在5个不同降水样点共采集30个土壤样品。将混合好的土壤样品各分为3份, 一份置于-80℃冰箱保存, 用于环境微生物基因组的提取, 一份置于4℃冰箱测试土壤样品铵态氮(NH4+-N)和硝态氮(NO3--N)含量, 剩余土壤样品, 用于土壤理化性质的测定。
1.3 土壤理化性质的测定
自然风干后的土壤样品, 过1 mm筛清除样品中可见的植物根系、凋落物、石子等杂物。采用烘干法测定土壤含水率; 电位法测定土壤pH (土壤与水混合比例为1∶5); 干烧法测定土壤全碳和总有机碳含量(Analytik Jena Multi N/C 3100, 德国); 高氯酸-硫酸消化法测定土壤全氮与全磷含量(SEA1 AutoAnalyzer3, 德国); 氯化钾(2 mol/L)浸提土壤硝态氮(NO3--N)和铵态氮(NH4+-N) (土壤与浸提溶液比例1∶4), 使用连续流动分析仪(SEA1 AutoAnalyzer3, 德国)测定其含量; 碳酸氢钠(0.5 mol/L)浸提土壤速效磷(土壤与浸提溶液比例1∶20), 使用连续流动分析仪(SEA1 AutoAnalyzer3, 德国)测定其含量。
1.4 16S rRNA基因扩增子测序
采用扩增子测序方法分析土壤细菌的多样性与群落结构。环境微生物基因组提取使用土壤DNA提取试剂盒(E.Z.N.A.® Soil DNA Kit, Omega Bio- Tek, 美国), 使用超微分光光度计(NANODROP 2000, Thermo SCIENTIFIC)检测核酸的质量和浓度。使用细菌V4区引物515F (5'-GTGCCAGCMGCC GCGG-3')和806R (5'-GGACTACHVGGGTWTCTA AT-3')进行PCR扩增和文库构建。PCR扩增条件为: 95℃预变性5 min; 95℃变性30 s, 56℃退火30 s, 72℃延伸45 s, 共30个循环; 72℃后延伸10 min。使用NEXTFLEX Rapid DNA-Seq Kit文库构建试剂盒制备测序文库; 使用Illumina NovaSeq PE250测序平台进行高通量测序; 下机后得到的序列首先使用FLASH软件对片段进行拼接, 允许片段重合长度为10 bp, 容错率为25%, 并使用Cutadapt (V1.9.1)去除低质量序列(< 150 bp); 截去Barcode及引物序列后, 进一步利用UPARSE软件对序列进行聚类(version 7.1,
1.5 低丰度细菌和高丰度细菌定义
依据样品ASV相对丰度, 参考Jiao和Lu (2020a)的方法, 将在所有样品中相对丰度 > 0.1%的ASVs定义为高丰度细菌类群, 相对丰度 < 0.01%的ASVs定义为低丰度细菌类群, 0.01% < 相对丰度< 0.1%之间的ASVs定义为中等丰度类群。高丰度细菌类群ASVs占总细菌ASVs的38.1%, 低丰度细菌类群ASVs占总细菌ASVs的55.1%。
1.6 数据分析
使用Shapiro-Wilk test对数据进行正态性检验,对于非正态数据进行对数转换, 使其接近正态分布, 然后使用单因素方差分析(one-way ANOVA)和LSD (least significant difference)多重比较分析降水差异对细菌群落α多样性、优势细菌门相对丰度及环境因子(除MAP以外)的影响。不同样点细菌β多样性的测定包括基于相对丰度的相异分布指数(Bray- Curtis dissimilarity)和基于物种相似性随地理距离的周转速率。使用非度量多维尺度排序分析(Non- metric multidimensional scaling, NMDS)、ANOSIM分析(analysis of similarities)和PERMANOVA (permutational multivariate analysis of variance)分析明确不同降水梯度细菌组成差异, 并利用非参数Kruskal-Wallis方法检验不同样点细菌群落β多样性差异。线性回归拟合细菌β多样性(Bray-Curtis distance)与样点间地理距离(Euclidean distance)之间的线性关系, 判断细菌β多样性随地理距离的周转速率。基于层次分割获取典范分析定量环境因子与地理距离对细菌群落组成的相对贡献率, 基于“packfor”包的向前选择去除环境因子冗余性。为了分析区域小范围降水差异对荒漠细菌群落生态系统功能或营养策略的影响, 使用细菌分类功能注释数据库(FAPROTAX 1.2) (Louca et al, 2016)来预测不同降水梯度土壤细菌群落的潜在功能。
基于物种最近系统发育距离指数βNTI和相对丰度Raup-Crick指数(RCbray)对不同降水样点的细菌群落构建框架进行分析(Vellend, 2010; Stegen 2013; Dini-Andreote et al, 2015; Ning et al, 2020)。该群落构建框架共分为5种模式: 同质选择(homogeneous selection, βNTI < -2)、同质扩散(homogenizing dispersal, |βNTI| < 2和RCbray < -0.95)、非主导过程(undominated, |βNTI| < 2和|RCbray| < 0.95)、扩散限制(dispersal limitation, |βNTI| < 2和|RCbray| > 0.95)和异质选择(heterogeneous selection, βNTI > 2)。由于系统发育保守性, 我们通过细菌群落间的平均最近物种距离(βMNTD)和最近物种指数(βNTI)计算样品间的系统发育组成变化(系统发育β多样性) (Vellend, 2010; Stegen et al, 2015)。使用Mantel检验分析土壤环境因子对微生物群落构建的影响。所有统计分析均基于R version 4.1完成, 所有图基于ggplot2包实现。
2 结果
2.1 不同降水样点的荒漠土壤理化特征
单因素方差分析显示, 不同降水样点荒漠土壤总碳具有显著差异(P < 0.05), 其中, SXS土壤(MAP 190 mm)的总碳含量(2.50 ± 0.22 g/kg)最高, CNN土壤(221 mm)总碳含量最低; 然而, 不同降水差异对荒漠土壤总有机碳(0.49‒0.92 g/kg)含量没有显著性的影响(P > 0.05); MAP > 204 mm的样点CNW (232 mm, pH 7.65 ± 0.05)、CNN (221 mm, pH 7.74 ± 0.07)和YZ (204 mm, pH 7.65 ± 0.14)的土壤pH显著低于SXS样点(190 mm, pH 8.00 ± 0.04) (P < 0.05) (附录1)。
2.2 荒漠土壤细菌多样性对区域降水差异的响应
区域降水差异对古尔班通古特沙漠总细菌和高丰度细菌类群的丰富度及Shannon多样性指数没有显著影响(P > 0.05, 附录11), 然而, 降水差异对低丰度细菌的丰富度及Shannon多样性指数均有显著影响(P < 0.05)。随降水减少, 降水量高的沙漠东部的CNW和CNN (MAP 232‒221 mm)低丰度细菌类群的物种丰富度显著高于降水量低的中西部的YZ、SXS和SXE 3个样点(MAP 181‒204 mm) (P < 0.05; 附录11)。
相似性分析显示, 不同样点总细菌、高丰度和低丰度细菌类群的组成间差异均显著大于组内差异(ANOSIM, R2 = 0.198‒1.000, P < 0.01; 图1a, 附录2)。多元置换方差分析进一步显示其区域降水差异对总细菌和不同丰度细菌的群落组成具有显著的影响(ADONIS, R2 = 0.139‒0.374, P < 0.05; 附录3), 低丰度细菌群落组成差异显著高于总细菌和高丰度细菌(图2)。基于Bray-Curtis的相异分布指数显示, YZ土壤不同丰度细菌群落扩散度低, 其β多样性显著高于SXS土壤(Kruskal-Wallis, P < 0.05, 图1b)。另外, 总细菌和不同丰度细菌类群物种组成的相似度均存在显著的距离衰减关系(distance-decay relationships, DDR) (P < 0.001; 图1c)。由空间距离衰减斜率可知, 低丰度细菌类群的物种空间周转速率最高(R2 = 0.014, Slope = -5.4 e-05, P < 0.001), 高丰度细菌类群的(R2 = 0.013, Slope = -4.7 e-05, P < 0.001)次之, 总细菌(R2 = 0.014, Slope = -2.2 e-05, P < 0.001)最低(图1c); β多样性分解结果显示, 总细菌、高丰度细菌和低丰度细菌类群沿降水梯度群落组成的差异以物种替换(species replacement)组分为主, 对β多样性的平均贡献分别为95.8%、89.3%和98.1%。
图1
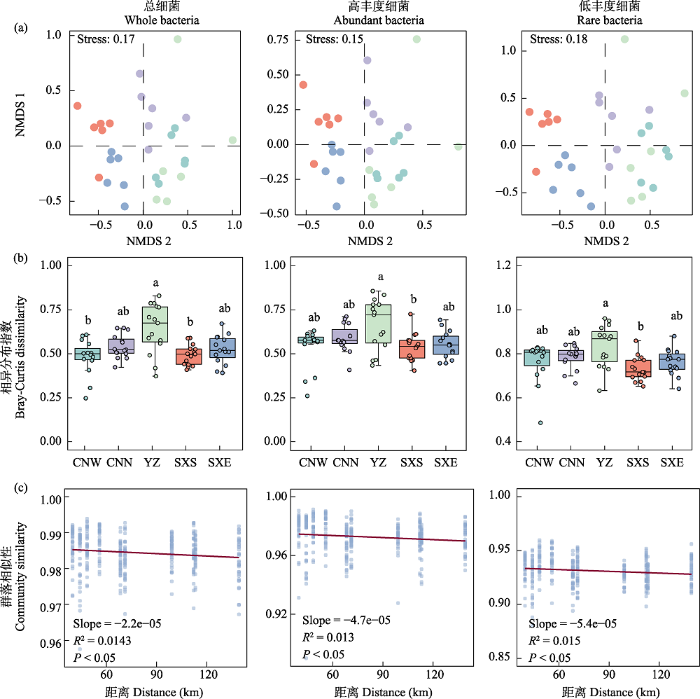
图1
区域降水差异对不同丰度细菌群落结构的影响。(a)基于Bray-Curtis距离的细菌群落结构非度量多维尺度排序。(b)降水对细菌群落Bray-Curtis相异分布指数的影响, 不同字母表示Bray-Curtis相异分布指数在不同降水梯度间的差异显著(P < 0.05)。(c)系统发育距离-衰减曲线显示群落相似性(1 ‒ βMNTD)与采样点之间地理距离的关系。CNW: 彩南西; CNN: 彩南北; YZ: 一站; SXS: 石西南; SXE: 石西东。
Fig. 1
The structure of bacterial communities with different abundances differed along the precipitation gradient. (a) Non-metric multidimensional scaling (NMDS) ordination based on Bray-Curtis similarity. (b) Effect of mean annual precipitation on the Bray-Curtis dissimilarity of bacterial communities, different letters indicate significant differences in Bray-Curtis dissimilarity between different precipitation gradients (P < 0.05). (c) Phylogenetic distance-decay curves showing community similarity (1 - βMNTD metric) against geographic distances between sampling sites.
图2
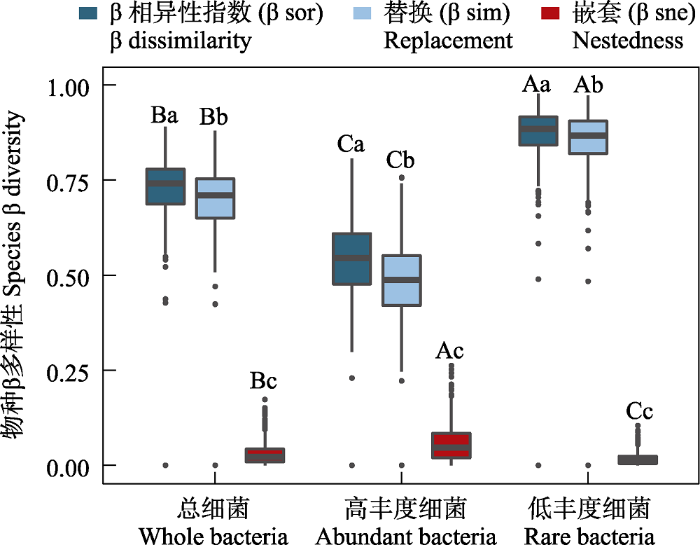
图2
不同丰度细菌β多样性及其物种替换和物种嵌套组分。不同大写字母表示同一丰度细菌类群β多样性组分间差异显著, 不同小写字母表示不同丰度细菌类群β多样性组分间差异显著(P < 0.05)。
Fig. 2
β diversity and its components (replacement and nestedness) of whole, abundant and rare bacterial community. Different capital letters indicate significant differences among β-diversity components of bacterial taxa of the same abundance. Different lowercase letters indicate significant differences among β-diversity components of whole, abundant and rare bacterial taxa (P < 0.05).
层次分割获取典范分析结果显示, 空间地理距离和气候因素是影响古尔班通古特沙漠细菌群落多样性的主要因素, 冗余分析显示MAP是影响荒漠细菌群落组成变异的关键环境因子(附录4, 附录12)。
基于FAPEOTAX功能预测结果, 总细菌类群一共注释了50种潜在功能, 高丰度类群细菌注释了36种, 低丰度细菌类群注释了49种。其中, 化能异养和有氧化能异养是荒漠不同丰度细菌类群的优势功能类群(62.6%‒66.3%), 随着区域MAP的减少, 这两类注释功能在CNN (39.7% ± 0.7%)和YZ (36.5% ± 1.9%)的功能OTUs相对丰度显著高于CNW (26.0% ± 3.3%)、SXS (32.4% ± 1.2%)、SXE (35.9% ± 1.6%) (附录10)。
2.3 细菌不同分类水平对降水差异的响应
放线菌门(19.1%‒28.6%)、变形菌门(18.8%‒ 26.4%)、绿弯菌门(11.1%‒20.1%)和拟杆菌门(8.9%‒ 11.2%)是荒漠表层土壤的优势细菌门。区域降水差异对放线菌门与拟杆菌门的相对丰度具有显著影响(P < 0.05), 其中, CNN (28.6% ± 1.8%)和SXS土壤(26.2% ± 1.2%)放线菌门的相对丰度显著高于CNW土壤(19.1% ± 1.8%) (P < 0.01), CNN (20.1% ± 1.6%)的绿弯菌门相对丰度显著高于CNW、YZ和SXS土壤(13.3% ± 1.4%) (P < 0.01) (附录7)。
优势细菌科对区域降水响应的趋势不同。古尔班通古特沙漠由西至东区域降水差异对优势红色杆菌科(8.2%‒3.8%)、AKIW781 (7.2%‒3.5%)和分枝杆菌科(2.4%‒3.7%)的相对丰度具有显著影响(P < 0.01), 对噬几丁质菌科(5.9%‒8.6%)、拜叶林克氏菌科(4.0%‒5.9%)、鞘脂单胞菌科(3.2%‒4.9%)和亚硝化螺菌科(4.5%‒1.5%)的相对丰富没有显著影响(P > 0.05)。另外, 绿弯菌门中未确定细菌AKIW781科是高丰度细菌与低丰度细菌类群中的优势细菌科, 其中, CNN土壤未确定细菌科AKIW781的相对丰度(7.2% ± 0.5%)最高, YZ土壤未确定细菌科AKIW781的相对丰度(3.1% ± 0.6%)最低(附录8)。
属水平分析表明, 区域降水差异对绿弯菌门未确定的细菌属WD210 (1.5%‒2.1%)、土壤红杆菌目未确定的细菌属67-14 (1.1%‒2.2%)、苔藓杆菌属(Bryobacter, 1.5%‒4%)、长微菌科未知属(Longimicrobium, 0.8%‒1.6%)和寡弯曲菌目未知属0319-6G20 (1.0%‒2.8%)的相对丰度具有显著影响(P < 0.05), 对未确定的绿弯菌门的TK10属(2.1%‒3.1%)、JG30-KF-CM45属(1.3%‒4.5%)、硝化螺旋菌属(Nitrospira, 0.3%‒7.0%)、未确定的放线菌纲的0319-7L14属(1.9%‒2.1%)、Chthoniobacter (0.4%‒3.6%)的相对丰度并没有显著影响。优势细菌属对区域降水响应的趋势不同, 长微菌科未知属(Longimicrobium)同是高丰度细菌与低丰度细菌类群中的优势细菌属(附录9)。
2.4 荒漠土壤细菌群落的构建模式及响应降水差异特征
荒漠土壤的群落构建分析显示, 不同丰度细菌类群均具有显著的系统发育信号(附录13)。结果表明, 扩散限制主导了古尔班通古特沙漠总细菌(73.0%)、高丰度细菌(79.0%)和低丰度细菌类群(52.0%)的群落构建过程; 然而, 相较于总细菌和高丰度细菌, 低丰度细菌群落构建过程同时受到强的异质选择作用(43.0%)。沿古尔班通古特沙漠降水梯度, 低丰度细菌类群的确定性过程先增加后降低, 确定性过程对沙漠东部CNN和中部YZ低丰度细菌群落构建很重要(图3)。
图3
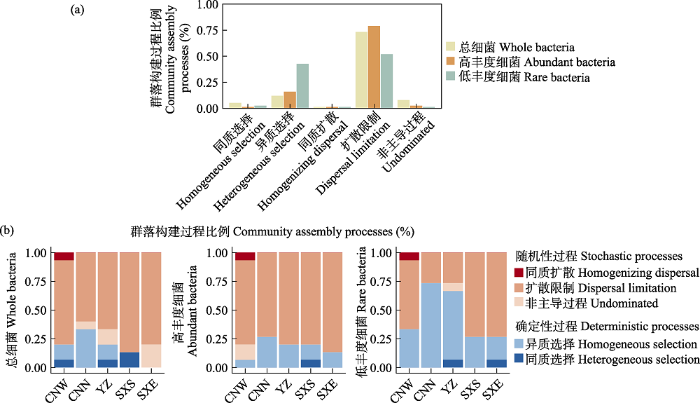
图3
整个样带不同丰度细菌群落构建(a)及沿降水梯度不同样点细菌群落构建过程的比例(b)。群落构建的比例包括确定性过程(同质选择和异质选择)以及随机过程(扩散限制和同质扩散)的支配, 以及不受任何单一过程支配的(“非主导过程”)。CNW: 彩南西; CNN: 彩南北; YZ: 一站; SXS: 石西南; SXE: 石西东。
Fig. 3
The fraction of assembly mechanism in whole, abundant and rare bacterial subcommunities based on the null model. The percent of turnover in whole, abundant and rare bacterial community assembly governed primarily by various deterministic, including homogeneous and heterogeneous selection, and stochastic processes, including dispersal limitations and homogenizing dispersal, as well as the fraction that was not dominated by any single process (‘Undominated’).
表1 不同丰度细菌群落βNTI与环境因子的Partial Mantel检验。粗体表示在P < 0.05水平上差异显著。
Table 1
| 变量 Variables | 控制变量 Controlled variables | R | P | |
|---|---|---|---|---|
| TP | Spatial + Environmental (excluding TP) | 0.03 | 0.633 | |
| 总细菌 Whole bacteria | Spatial | Environmental (excluding TP) + TP | 0.26 | 0.002** |
| Environmental (excluding TP) | Spatial + TP | 0.06 | 0.226 | |
| MAP | Spatial + Environmental (excluding MAP) | 0.357 | 0.001*** | |
| 高丰度细菌 Abundant bacteria | Spatial | Spatial + Environmental (excluding MAP) + MAP | 0.361 | 0.001*** |
| Environmental (excluding MAP) | Spatial + MAP | 0.029 | 0.322 | |
| MAP | Spatial + Environmental (excluding MAP) | 0.474 | 0.001*** | |
| TP | Spatial + Environmental (excluding TP) | 0.012 | 0.395 | |
| 低丰度细菌 Rare bacteria | Spatial | Spatial + Environmental (excluding MAP) + MAP | 0.502 | 0.001*** |
| Environmental (excluding MAP + TP) | Spatial + MAP + TP | 0.014 | 0.369 |
图5
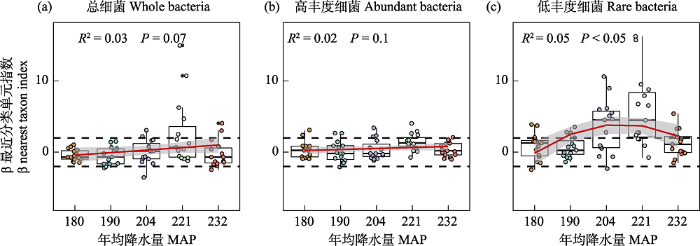
图5
降水介导了不同丰度细菌群落构建中确定性和随机性过程相对重要性。水平虚线表示βNTI显著性阈值为+2和-2。
Fig. 5
Precipitation balances the relative importance of deterministic and stochastic in the community assembly of whole, abundant and rare bacterial communities. Horizontal-dashed lines indicate the βNTI significance thresholds of +2 and −2.
3 讨论
3.1 荒漠土壤细菌多样性对区域降水变化的响应
长期历史性降水差异对塑造不同生态系统土壤的细菌群落结构具有重要作用。在全球尺度和大陆尺度, 降水差异所驱动的细菌群落多样性分布与土壤养分循环及多功能性密切相关, 在很大程度上也决定生态系统的生态功能(Delgado-Baquerizo et al, 2016)。尽管在大量模拟控制实验以及在大尺度自然降水梯度中, 降水差异驱动不同生态系统, 包括森林、草原和农田土壤细菌群落的数量与多样性(Richter-Heitmann et al, 2020; Yang et al, 2021; Zhou et al, 2021), 但对于较小范围历史性降水差异对荒漠土壤细菌分布具有怎样的影响, 仍然了解得较少。本研究基于古尔班通古特沙漠不同降水样点, 初步证明了区域小范围降水差异与荒漠细菌的多样性和群落组成具有密切的联系, 除空间地理距离外, MAP是影响荒漠细菌群落组成变异的关键环境因子。古尔班通古特沙漠区域范围年均降水差异(介于181‒232 mm)较小, 不同降水梯度样点间细菌群落组成均具有显著差异(图1b, 图2, 附录4), 这与在大尺度自然降水梯度, 如中国北方草原生态系统(MAP 159‒516 mm) (Yang et al, 2022)、以色列干旱半干旱生态系统(MAP 100‒450 mm) (Peay et al, 2017)以及内蒙古干旱半干旱草原(MAP 167‒333 mm) (Yao et al, 2017)细菌群落响应降水差异的趋势较一致, 推测区域较小降水差异显著影响荒漠土壤的细菌群落组成, 可能与荒漠生态系统主要受到水分的限制紧密相关。对干旱荒漠土壤, 区域范围内小频率降水差异引起的水分与养分脉冲效应的差异, 会使微生物多样性和群落组成发生变化(Steven et al, 2021)。另外, 由于受到水分及养分限制, 荒漠土壤中多数细菌通常处于休眠状态, 区域小范围降水的差异仍然能够促使细菌复苏差异, 一些死细菌细胞的裂解也会导致土壤异质性增加。最后, 降水差异促使荒漠本身难分解的有机碳矿化差异, 进一步影响细菌群落的分布(Throop et al, 2020)。
不同丰度荒漠细菌类群差异响应区域降水变化。相较于低丰度细菌群落, 总细菌和高丰度细菌多样性和群落组成对降水差异表现出一定的稳定性, 而低丰度细菌群落多样性及群落组成敏感响应小范围降水变化(MAP介于180‒230 mm)。例如, 降水量相对较高的CNW和CNN (MAP 230‒220 mm)土壤, 其低丰度细菌类群的物种丰富度显著高于降水量低的YZ、SXS和SXE (MAP 181.0‒204.3 mm), 而总细菌和高丰度细菌的物种丰富度及多样性在不同样点间无显著差异(Kruskal-Wallis, P < 0.05, 附录6)。此外, 低丰度细菌群落变异以及空间物种替换大于总细菌和高丰度细菌类群(图2, 附录2), 相较于高丰度细菌, 低丰度细菌对降水变化的响应更敏感可能有两个原因: (1)高丰度和低丰度细菌环境适应力存在差异, 低丰度细菌群落具有较窄的生态位宽度, 更容易受到环境过滤作用, 迅速响应环境变化以适应变化的新环境(Gao et al, 2020; Jiao & Lu, 2020a); (2)低丰度细菌类群对资源竞争能力有限, 仅能够在有限的资源条件下存活, 所以对降水变化更敏感响应(Delgado-Baquerizo et al, 2018; Shu et al, 2021)。此外, 相较于高丰度类群主要包含化能异养和有氧化能异养功能类群, 低丰度细菌还包含了更丰富的具有芳香化合物降解、尿素分解、发酵途径等功能类群, 并且小范围降水变化对低丰度细菌优势功能类群的相对丰度均产生了不同程度的影响, 这表明低丰度细菌类群具有更丰富的功能类型, 其对小范围降水变化的响应更加敏感, 小范围降水对不同丰度细菌功能类群的影响可能会进一步影响生态系统的功能。土壤微生物对气候变化下的温带荒漠土壤多功能有着显著的反馈作用(Hu et al, 2021), 我们认为这种区域范围较小的降水差异能够显著影响细菌群落多样性与其物种周转, 促使我们进一步认识水分对荒漠微生物响应环境变化模式的重要作用。
3.2 荒漠土壤细菌多样性维持机制及响应降水变化的特征
全球变化背景下, 中亚干旱区荒漠面积持续扩大, 小范围降水变化是如何影响荒漠土壤细菌群落的构建机制仍然不清楚。传统观点认为, 环境变化幅度的增加会提高微生物群落构建过程中确定性过程相对于随机过程的重要性(Friedmann & Galun, 1974), 而基于古尔班通古特沙漠区域降水差异的研究结果发现, 荒漠表层土壤细菌群落构建由确定性过程和随机性过程共同决定, 其中, 随机过程主导了荒漠表层土壤细菌的群落构建过程(图2)。一方面, 荒漠生态系统生境相对单一, 相较于多由确定性过程主导的复杂生态系统, 如森林和农田的微生物群落构建(Jiao & Lu, 2020a, b; Liu L et al, 2021), 随机作用(扩散限制)在维持荒漠区域降水梯度下土壤细菌群落多样性非常重要, 扩散事件可以改变细菌群落形成的速度, 从而改变细菌群落的组成, 增加细菌群落的β多样性, 先前在荒漠草原降水样带的研究也证实了这一结论(Yang et al, 2022), 值得注意的是, 对比总细菌(73.0%), 扩散限制对高丰度细菌群落构建(79.0%)的主导作用更强, 表明即使在区域荒漠, 高丰度细菌类群仍然表现出了宽的生态位适应特征, 这可能与高丰度细菌“资源获取型”的生态策略有关, 从而具有更强的空间与资源竞争力, 以及更强的环境胁迫耐受性(Gao et al, 2020; Jiao & Lu, 2020a; Wan et al, 2021)。另一方面, 荒漠地区常年伴有风沙和尘暴, 其荒漠表层土壤微生物可实现远距离的扩散传播, 很大程度上也增加了随机作用 (Liu NN et al, 2021), 此外细菌的生活史和生活策略可能也介导了荒漠细菌的扩散作用, 如放线菌门(19.1%‒28.6%)始终是古尔班通古特沙漠不同丰度细菌类群的优势细菌门(附录7), 这可能与放线菌产孢具有更强的扩散和耐干旱能力有关(Mohammadipanah & Wink, 2016)。
相较于总细菌和高丰度细菌, 低丰度细菌群落构建同时受到强的扩散限制(52.0%)和异质选择作用(42.7%), 推测区域降水差异直接或间接地增强了荒漠土壤异质性对低丰度细菌类群的环境过滤作用。Jiao和Lu (2020a)发现, 农田土壤低丰度细菌群落的构建过程更多地由随机性过程主导, 异质选择对农田与荒漠土壤细菌群落构建影响的差异, 可能与其土壤环境差异大有关, 长期灌溉、施肥和作物种植等促使农田更偏向同质生境, 荒漠生态系统土壤养分贫瘠, 持水力差和抗干扰能力弱, 长期激烈的水分和养分竞争促使低丰度细菌类群多为特化优势种, 更容易受到环境过滤作用的影响(徐鹏等, 2022)。此外, 我们的研究表明, 荒漠土壤低丰度细菌多样性的维持更依赖于降水变化(表1, 图4)。在大多数自然生境中, 低丰度细菌类群具有更窄的生态位以及更低的资源竞争能力(Jiao et al, 2017), 尽管由古尔班通古特沙漠西部至东部, 其MAP差异仅约50 mm, 然而MAP强烈介导了低丰度细菌类群的随机性过程与确定性过程之间的平衡, 随荒漠土壤MAP增加, 荒漠低丰度细菌群落构建过程由随机性过程转向确定性过程主导, 当MAP约大于200 mm, 低丰度细菌类群的群落构建过程的主导作用再转变为随机性作用(图5)。推测在荒漠区域尺度,较高频率的小范围降水事件不仅驱动荒漠土壤的水分, 且其养分脉冲也很大程度上改变了细菌多样性及其养分循环。最近的研究也表明, 在半干旱区, 植物多样性及其生产力是影响生态系统功能与稳定性的关键限制性因素, 而在干旱区与极端干旱区, 土壤微生物多样性则是影响生态系统功能与稳定性的关键限制性因素(Hu et al, 2021), 因此, 古尔班通古特沙漠区域小范围降水差异影响土壤细菌群落多样性, 可能将进一步影响荒漠的土壤生态系统功能; 另外, 低丰度细菌β多样性敏感响应区域小范围降水差异, 且在不同降水梯度下表现出非线性调控模式, 一方面有利于干旱环境荒漠细菌通过快速调整其生态位或物种相互作用, 协同可利用养分, 另一方面, 低丰度细菌依赖于降水对指示荒漠化的土壤细菌多样性和功能冗余具有重要作用。
图4
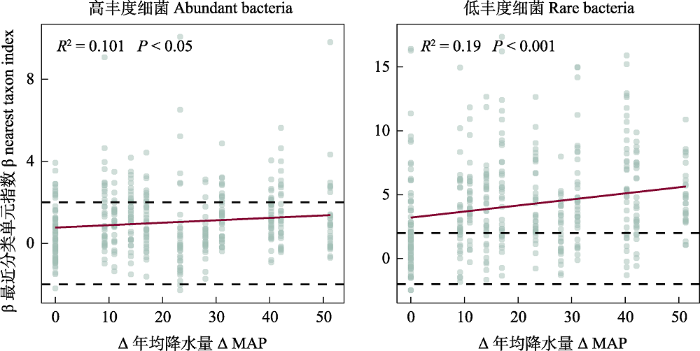
图4
细菌β最近分类单元指数βNTI与年均降水量之间的Spearman相关性。ΔMAP表示样点和样点之间年均降水量的差异。
Fig. 4
Relationships between (β nearest taxon index, βNTI) and differences in MAP (mean annual precipitation) for the abundant and rare bacterial communities. ΔMAP indicate the differences of MAP between sample sites.
附录 Supplementary Material
附录1 古尔班通古特沙漠样点地理、气候和土壤理化特性
Appendix 1 Geographic, climate, soil physicochemical properties of sampling sites in the Gurbantunggut Desert
附录2 相似性分析(ANOSIM)检验降水对荒漠细菌群落组成的影响
Appendix 2 Analysis of similarity (ANOSIM) statistics testing effects of mean annual precipitation (MAP) on bacterial community composition
附录3 置换多元方差分析(PERMANOVA)检验降水对细菌群落的影响
Appendix 3 Permutational multivariate analysis of variance (PERMANOVA) statistics testing effects of mean annual precipitation (MAP) on bacterial communities
附录4 冗余分析筛选影响细菌β多样性在降水梯度上的环境因子
Appendix 4 Redundancy analysis to screen the environmental factors affecting bacterial β diversity along the precipitation gradient
附录5 不同丰度细菌群落βNTI与环境因子的Mantel test
Appendix 5 Mantel tests of environmental variables against the phylogenetic turnover (β nearest taxon index) of whole, abundant and rare bacterial subcommunities in the Gurbantunggut Desert
附录6 研究区域位置及采样点示意图
Appendix 6 Schematic diagram of the location and sampling points of the study area
附录7 沿降水梯度细菌前10个门的相对丰度
Appendix 7 Relative abundances of the top 10 bacteria phylum with precipitation in the Gurbantunggut Desert
附录8 沿降水梯度细菌前10个科的相对丰度
Appendix 8 Relative abundances of the top 10 bacterial family with precipitation in the Gurbantunggut Desert.
附录9 沿降水梯度细菌前10优势细菌属的相对丰度
Appendix 9 Relative abundances of the top 10 bacteria genera with precipitation in the Gurbantunggut Desert
附录10 FAPROTAX功能预测的丰度前10的优势功能类群
Appendix 10 FAPROTAX functional prediction analysis predicts the top 10 dominant bacterial functions
附录11 降水对不同丰度细菌群落α多样性的影响
Appendix 11 Effect of mean annual precipitation on the α-diversity of whole, abundant and rare bacteria communities
附录12 层次分割获取典范分析解释变量相对重要性
Appendix 12 Hierarchical partitioning for canonical correspondence analysis and redundancy analysis plots of relative importance of explanatory variables
附录13 细菌最优环境欧氏距离和系统发育距离的Mantel相关图
Appendix 13 Mantel correlogram for pairwise Euclidean distances of bacterial ASVs’ optimal environmental conditions and phylogenetic distances
参考文献
Soil microbial abundance and diversity along a low precipitation gradient
DOI:10.1007/s00248-010-9727-1
PMID:20683588
[本文引用: 1]

The exploration of spatial patterns of abundance and diversity patterns along precipitation gradients has focused for centuries on plants and animals; microbial profiles along such gradients are largely unknown. We studied the effects of soil pH, nutrient concentration, salinity, and water content on bacterial abundance and diversity in soils collected from Mediterranean, semi-arid, and arid sites receiving approximately 400, 300, and 100 mm annual precipitation, respectively. Bacterial diversity was evaluated by terminal restriction fragment length polymorphism and clone library analyses and the patterns obtained varied with the climatic regions. Over 75% of the sequenced clones were unique to their environment, while ∼2% were shared by all sites, yet, the Mediterranean and semi-arid sites had more common clones (∼9%) than either had with the arid site (4.7% and 6%, respectively). The microbial abundance, estimated by phospholipid fatty acids and real-time quantitative PCR assays, was significantly lower in the arid region. Our results indicate that although soil bacterial abundance decreases with precipitation, bacterial diversity is independent of precipitation gradient. Furthermore, community composition was found to be unique to each ecosystem.
Environmental filtering process has more important roles than dispersal limitation in shaping large-scale prokaryotic beta diversity patterns of grassland soils
DOI:10.1007/s00248-016-0762-4
PMID:27072664
[本文引用: 1]
Despite the utmost importance of microorganisms in maintaining ecosystem functioning and their ubiquitous distribution, our knowledge of the large-scale pattern of microbial diversity is limited, particularly in grassland soils. In this study, the microbial communities of 99 soil samples spanning over 3000 km across grassland ecosystems in northern China were investigated using high-throughput sequencing to analyze the beta diversity pattern and the underlying ecological processes. The microbial communities were dominated by Proteobacteria, Actinobacteria, Acidobacteria, Chloroflexi, and Planctomycetes across all the soil samples. Spearman's correlation analysis indicated that climatic factors and soil pH were significantly correlated with the dominant microbial taxa, while soil microbial richness was positively linked to annual precipitation. The environmental divergence-dissimilarity relationship was significantly positive, suggesting the importance of environmental filtering processes in shaping soil microbial communities. Structural equation modeling found that the deterministic process played a more important role than the stochastic process on the pattern of soil microbial beta diversity, which supported the predictions of niche theory. Partial mantel test analysis have showed that the contribution of independent environmental variables has a significant effect on beta diversity, while independent spatial distance has no such relationship, confirming that the deterministic process was dominant in structuring soil microbial communities. Overall, environmental filtering process has more important roles than dispersal limitation in shaping microbial beta diversity patterns in the grassland soils.
Change and analysis of annual desertification and climate factors in Inner Mongolia using MODIS data
内蒙古荒漠化年际动态变化及与气候因子分析
Dispersal limitation relative to environmental filtering governs the vertical small-scale assembly of soil microbiomes during restoration
DOI:10.1111/jpe.v57.2 URL [本文引用: 1]
Microbial diversity drives multifunctionality in terrestrial ecosystems
DOI:10.1038/ncomms10541
PMID:26817514
[本文引用: 1]
Despite the importance of microbial communities for ecosystem services and human welfare, the relationship between microbial diversity and multiple ecosystem functions and services (that is, multifunctionality) at the global scale has yet to be evaluated. Here we use two independent, large-scale databases with contrasting geographic coverage (from 78 global drylands and from 179 locations across Scotland, respectively), and report that soil microbial diversity positively relates to multifunctionality in terrestrial ecosystems. The direct positive effects of microbial diversity were maintained even when accounting simultaneously for multiple multifunctionality drivers (climate, soil abiotic factors and spatial predictors). Our findings provide empirical evidence that any loss in microbial diversity will likely reduce multifunctionality, negatively impacting the provision of services such as climate regulation, soil fertility and food and fibre production by terrestrial ecosystems.
A global atlas of the dominant bacteria found in soil
DOI:10.1126/science.aap9516
PMID:29348236
[本文引用: 2]
The immense diversity of soil bacterial communities has stymied efforts to characterize individual taxa and document their global distributions. We analyzed soils from 237 locations across six continents and found that only 2% of bacterial phylotypes (~500 phylotypes) consistently accounted for almost half of the soil bacterial communities worldwide. Despite the overwhelming diversity of bacterial communities, relatively few bacterial taxa are abundant in soils globally. We clustered these dominant taxa into ecological groups to build the first global atlas of soil bacterial taxa. Our study narrows down the immense number of bacterial taxa to a "most wanted" list that will be fruitful targets for genomic and cultivation-based efforts aimed at improving our understanding of soil microbes and their contributions to ecosystem functioning.Copyright © 2018 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. No claim to original U.S. Government Works.
Disentangling mechanisms that mediate the balance between stochastic and deterministic processes in microbial succession
Distinct community assembly processes of abundant and rare soil bacteria in coastal wetlands along an inundation gradient
Beyond biogeographic patterns: Processes shaping the microbial landscape
DOI:10.1038/nrmicro2795
PMID:22580365
[本文引用: 1]
Recently, microbiologists have established the existence of biogeographic patterns among a wide range of microorganisms. The focus of the field is now shifting to identifying the mechanisms that shape these patterns. Here, we propose that four processes - selection, drift, dispersal and mutation - create and maintain microbial biogeographic patterns on inseparable ecological and evolutionary scales. We consider how the interplay of these processes affects one biogeographic pattern, the distance-decay relationship, and review evidence from the published literature for the processes driving this pattern in microorganisms. Given the limitations of inferring processes from biogeographic patterns, we suggest that studies should focus on directly testing the underlying processes.
Aridity-driven shift in biodiversity-soil multifunctionality relationships
DOI:10.1038/s41467-021-25641-0
PMID:34504089
[本文引用: 2]
Relationships between biodiversity and multiple ecosystem functions (that is, ecosystem multifunctionality) are context-dependent. Both plant and soil microbial diversity have been reported to regulate ecosystem multifunctionality, but how their relative importance varies along environmental gradients remains poorly understood. Here, we relate plant and microbial diversity to soil multifunctionality across 130 dryland sites along a 4,000 km aridity gradient in northern China. Our results show a strong positive association between plant species richness and soil multifunctionality in less arid regions, whereas microbial diversity, in particular of fungi, is positively associated with multifunctionality in more arid regions. This shift in the relationships between plant or microbial diversity and soil multifunctionality occur at an aridity level of ∼0.8, the boundary between semiarid and arid climates, which is predicted to advance geographically ∼28% by the end of the current century. Our study highlights that biodiversity loss of plants and soil microorganisms may have especially strong consequences under low and high aridity conditions, respectively, which calls for climate-specific biodiversity conservation strategies to mitigate the effects of aridification.© 2021. The Author(s).
Soil microbiomes and climate change
DOI:10.1038/s41579-019-0265-7
PMID:31586158
[本文引用: 1]
The soil microbiome governs biogeochemical cycling of macronutrients, micronutrients and other elements vital for the growth of plants and animal life. Understanding and predicting the impact of climate change on soil microbiomes and the ecosystem services they provide present a grand challenge and major opportunity as we direct our research efforts towards one of the most pressing problems facing our planet. In this Review, we explore the current state of knowledge about the impacts of climate change on soil microorganisms in different climate-sensitive soil ecosystems, as well as potential ways that soil microorganisms can be harnessed to help mitigate the negative consequences of climate change.
Community assembly processes of the microbial rare biosphere
DOI:S0966-842X(18)30047-7
PMID:29550356
[本文引用: 1]
Our planet teems with microorganisms that often present a skewed abundance distribution in a local community, with relatively few dominant species coexisting alongside a high number of rare species. Recent studies have demonstrated that these rare taxa serve as limitless reservoirs of genetic diversity, and perform disproportionate types of functions despite their low abundances. However, relatively little is known about the mechanisms controlling rarity and the processes promoting the development of the rare biosphere. Here, we propose the use of multivariate cut-offs to estimate rare species and phylogenetic null models applied to predefined rare taxa to disentangle the relative influences of ecoevolutionary processes mediating the assembly of the rare biosphere. Importantly, the identification of the factors controlling rare species assemblages is critical for understanding the types of rarity, how the rare biosphere is established, and how rare microorganisms fluctuate over spatiotemporal scales, thus enabling prospective predictions of ecosystem responses.Copyright © 2018 Elsevier Ltd. All rights reserved.
Biogeography and ecological diversity patterns of rare and abundant bacteria in oil-contaminated soils
DOI:10.1111/mec.14218
PMID:28665016
[本文引用: 1]
Revealing the biogeographies and ecologies of rare and abundant microorganisms is crucial to understand ecosystem diversity and function. In this study, we investigated the biogeographic assemblies and ecological diversity patterns of rare and abundant bacteria in long-term oil-contaminated soils at intervals of 46-360 km by performing high-throughput sequencing of 16S rRNA genes. The results clearly revealed distinct distribution patterns for rare and abundant bacteria in soil samples. Rare taxa were unevenly distributed; however, abundant taxa were ubiquitous across all samples. Both rare and abundant subcommunities showed significant distance-decay relationships, and their assemblies were driven by different factors. The rare subcommunity primarily exhibited a spatially structured distribution (i.e., stochastic processes), while edaphic factors (i.e., deterministic processes) largely contributed to the structure of the abundant subcommunity. A network analysis revealed closer relationships between abundant bacteria and their heightened influence on other co-occurrences in the community compared with rare species. In conclusion, rare microbial taxa may play potential roles in maintaining ecosystem diversity, although they do not appear to be central to microbial networks. Abundant microbes are vital for microbial co-occurrences in oil-contaminated soils, and high relative abundance and ubiquitous distribution suggest potential roles in the degradation of organic pollutants.© 2017 John Wiley & Sons Ltd.
Abundant fungi adapt to broader environmental gradients than rare fungi in agricultural fields
DOI:10.1111/gcb.v26.8 URL [本文引用: 7]
Soil pH and temperature regulate assembly processes of abundant and rare bacterial communities in agricultural ecosystems
DOI:10.1111/emi.v22.3 URL [本文引用: 3]
Changes in community assembly may shift the relationship between biodiversity and ecosystem function
DOI:10.3389/fmicb.2014.00424 PMID:25165465 [本文引用: 1]
Differentiation strategies of soil rare and abundant microbial taxa in response to changing climatic regimes
DOI:10.1111/1462-2920.14945
PMID:32067386
Despite the important roles of soil microbes, especially the most diverse rare taxa in maintaining community diversity and multifunctionality, how different climate regimes alter the stability and functions of the rare microbial biosphere remains unknown. We reciprocally transplanted field soils across a latitudinal gradient to simulate climate change and sampled the soils annually after harvesting the maize over the following 6 years (from 2005 to 2011). By sequencing microbial 16S ribosomal RNA gene amplicons, we found that changing climate regimes significantly altered the composition and dynamics of soil microbial communities. A continuous succession of the rare and abundant communities was observed. Rare microbial communities were more stable under changing climatic regimes, with lower variations in temporal dynamics, and higher stability and constancy of diversity. More nitrogen cycling genes were detected in the rare members than in the abundant members, including amoA, napA, nifH, nirK, nirS, norB and nrfA. Random forest analysis and receiver operating characteristics analysis showed that rare taxa may act as potential contributors to maize yield under changing climatics. The study indicates that the taxonomically and functionally diverse rare biosphere has the potential to increase functional redundancy and enhance the ability of soil communities to counteract environmental disturbances. With ongoing global climate change, exploring the succession process and functional changes of rare taxa may be important in elucidating the ecosystem stability and multifunctionality that are mediated by microbial communities.© 2020 Society for Applied Microbiology and John Wiley & Sons Ltd.
Diversity of marine heatwaves in the South China Sea regulated by ENSO phase
DOI:10.1175/JCLI-D-21-0309.1
URL
[本文引用: 1]
Marine heatwaves (MHWs) in the South China Sea (SCS) have dramatic impacts on local ecosystems, fisheries, and aquacultures. Our results show that SCS MHWs were strongly regulated by El Niño–Southern Oscillation (ENSO) with a distinct life cycle during 1982–2018. Based on the ENSO-associated sea surface temperature anomaly (SSTA) warming peaks in the SCS, we can classify SCS MHWs into three categories: El Niño-P1 during the first warming peak of El Niño from September to the following February, El Niño-P2 during the second warming peak of El Niño from the following June to September, and La Niña-P1 during the single warming peak of La Niña from the following February to May. The three types of SCS MHWs are all affected by the lower-level enhanced anticyclone over the western North Pacific (WNP), but their physical mechanisms are quite different. In El Niño-P1, SCS MHWs are mostly induced by enhanced net downward shortwave radiation and reduced latent heat flux loss over the southwestern and northern SCS, respectively. In El Niño-P2, SCS MHWs are primarily attributed to weaker entrainment cooling caused by a local enhanced anticyclone and stronger Ekman downwelling in the central-northern SCS. However, in La Niña-P1, SCS MHWs are mainly contributed by the reduced latent heat loss due to the weaker WNP anticyclone centered east of the Philippines on the pentad time scale. The distinct spatial distributions of MHWs show phase locking with ENSO-associated SCS SSTA warming, which provides a potential seasonal forecast of SCS MHWs according to the ENSO phase.
Changes in assembly processes of soil microbial communities during secondary succession in two subtropical forests
DOI:10.1016/j.soilbio.2021.108144 URL [本文引用: 1]
The biogeography of abundant and rare bacterioplankton in the lakes and reservoirs of China
DOI:10.1038/ismej.2015.29 [本文引用: 1]
Relative importance of deterministic and stochastic processes on soil microbial community assembly in temperate grasslands
DOI:10.3390/microorganisms9091929
URL
[本文引用: 1]
Changes in species composition across communities, i.e., β-diversity, is a central focus of ecology. Compared to macroorganisms, the β-diversity of soil microbes and its drivers are less studied. Whether the determinants of soil microbial β-diversity are consistent between soil depths and between abundant and rare microorganisms remains controversial. Here, using the 16S-rRNA of soil bacteria and archaea sampled at different soil depths (0–10 and 30–50 cm) from 32 sites along an aridity gradient of 1500 km in the temperate grasslands in northern China, we compared the effects of deterministic and stochastic processes on the taxonomic and phylogenetic β-diversity of soil microbes. Using variation partitioning and null models, we found that the taxonomic β-diversity of the overall bacterial communities was more strongly determined by deterministic processes in both soil layers (the explanatory power of environmental distance in topsoil: 25.4%; subsoil: 47.4%), while their phylogenetic counterpart was more strongly determined by stochastic processes (the explanatory power of spatial distance in topsoil: 42.1; subsoil 24.7%). However, in terms of abundance, both the taxonomic and phylogenetic β-diversity of the abundant bacteria in both soil layers was more strongly determined by deterministic processes, while those of rare bacteria were more strongly determined by stochastic processes. In comparison with bacteria, both the taxonomic and phylogenetic β-diversity of the overall abundant and rare archaea were strongly determined by deterministic processes. Among the variables representing deterministic processes, contemporary and historical climate and aboveground vegetation dominated the microbial β-diversity of the overall and abundant microbes of both domains in topsoils, but soil geochemistry dominated in subsoils. This study presents a comprehensive understanding on the β-diversity of soil microbial communities in the temperate grasslands in northern China. Our findings highlight the importance of soil depth, phylogenetic turnover, and species abundance in the assembly processes of soil microbial communities.
Decoupling function and taxonomy in the global ocean microbiome
DOI:10.1126/science.aaf4507
PMID:27634532
[本文引用: 1]
Microbial metabolism powers biogeochemical cycling in Earth's ecosystems. The taxonomic composition of microbial communities varies substantially between environments, but the ecological causes of this variation remain largely unknown. We analyzed taxonomic and functional community profiles to determine the factors that shape marine bacterial and archaeal communities across the global ocean. By classifying >30,000 marine microorganisms into metabolic functional groups, we were able to disentangle functional from taxonomic community variation. We find that environmental conditions strongly influence the distribution of functional groups in marine microbial communities by shaping metabolic niches, but only weakly influence taxonomic composition within individual functional groups. Hence, functional structure and composition within functional groups constitute complementary and roughly independent "axes of variation" shaped by markedly different processes.Copyright © 2016, American Association for the Advancement of Science.
Regional differences in plant diversity in the southern Gurbantonggut Desert
古尔班通古特沙漠南部植物多样性的区域差异
Actinobacteria from arid and desert habitats: Diversity and biological activity
Differences in precipitation regime shape microbial community composition and functional potential in Namib Desert soils
DOI:10.1007/s00248-021-01785-w [本文引用: 1]
Significant impacts of increasing aridity on the arid soil microbiome
Soil pH determines bacterial distribution and assembly processes in natural mountain forests of Eastern China
DOI:10.1111/geb.v30.11 URL [本文引用: 1]
A quantitative framework reveals ecological drivers of grassland microbial community assembly in response to warming
DOI:10.1038/s41467-020-18560-z
PMID:32948774
[本文引用: 1]
Unraveling the drivers controlling community assembly is a central issue in ecology. Although it is generally accepted that selection, dispersal, diversification and drift are major community assembly processes, defining their relative importance is very challenging. Here, we present a framework to quantitatively infer community assembly mechanisms by phylogenetic bin-based null model analysis (iCAMP). iCAMP shows high accuracy (0.93-0.99), precision (0.80-0.94), sensitivity (0.82-0.94), and specificity (0.95-0.98) on simulated communities, which are 10-160% higher than those from the entire community-based approach. Application of iCAMP to grassland microbial communities in response to experimental warming reveals dominant roles of homogeneous selection (38%) and 'drift' (59%). Interestingly, warming decreases 'drift' over time, and enhances homogeneous selection which is primarily imposed on Bacillales. In addition, homogeneous selection has higher correlations with drought and plant productivity under warming than control. iCAMP provides an effective and robust tool to quantify microbial assembly processes, and should also be useful for plant and animal ecology.
Aridity threshold induces abrupt change of soil abundant and rare bacterial biogeography in dryland ecosystems
Convergence and contrast in the community structure of bacteria, fungi and archaea along a tropical elevation-climate gradient
Responses of soil total microbial biomass and community compositions to rainfall reductions
DOI:10.1016/j.soilbio.2017.09.028 URL [本文引用: 1]
Stochastic dispersal rather than deterministic selection explains the spatio-temporal distribution of soil bacteria in a temperate grassland
DOI:10.3389/fmicb.2020.01391
PMID:32695081
[本文引用: 1]
Spatial and temporal processes shaping microbial communities are inseparably linked but rarely studied together. By Illumina 16S rRNA sequencing, we monitored soil bacteria in 360 stations on a 100 square meter plot distributed across six intra-annual samplings in a rarely managed, temperate grassland. Using a multi-tiered approach, we tested the extent to which stochastic or deterministic processes influenced the composition of local communities. A combination of phylogenetic turnover analysis and null modeling demonstrated that either homogenization by unlimited stochastic dispersal or scenarios, in which neither stochastic processes nor deterministic forces dominated, explained local assembly processes. Thus, the majority of all sampled communities (82%) was rather homogeneous with no significant changes in abundance-weighted composition. However, we detected strong and uniform taxonomic shifts within just nine samples in early summer. Thus, community snapshots sampled from single points in time or space do not necessarily reflect a representative community state. The potential for change despite the overall homogeneity was further demonstrated when the focus shifted to the rare biosphere. Rare OTU turnover, rather than nestedness, characterized abundance-independent β-diversity. Accordingly, boosted generalized additive models encompassing spatial, temporal and environmental variables revealed strong and highly diverse effects of space on OTU abundance, even within the same genus. This pure spatial effect increased with decreasing OTU abundance and frequency, whereas soil moisture - the most important environmental variable - had an opposite effect by impacting abundant OTUs more than the rare ones. These results indicate that - despite considerable oscillation in space and time - the abundant and resident OTUs provide a community backbone that supports much higher β-diversity of a dynamic rare biosphere. Our findings reveal complex interactions among space, time, and environmental filters within bacterial communities in a long-established temperate grassland.Copyright © 2020 Richter-Heitmann, Hofner, Krah, Sikorski, Wüst, Bunk, Huang, Regan, Berner, Boeddinghaus, Marhan, Prati, Kandeler, Overmann and Friedrich.
Rare prokaryotic sub-communities dominate the complexity of ecological networks and soil multinutrient cycling during long-term secondary succession in China’s Loess Plateau
DOI:10.1016/j.scitotenv.2021.145737 URL [本文引用: 1]
Groundwater-surface water mixing shifts ecological assembly processes and stimulates organic carbon turnover
DOI:10.1038/ncomms11237
PMID:27052662
[本文引用: 1]
Environmental transitions often result in resource mixtures that overcome limitations to microbial metabolism, resulting in biogeochemical hotspots and moments. Riverine systems, where groundwater mixes with surface water (the hyporheic zone), are spatially complex and temporally dynamic, making development of predictive models challenging. Spatial and temporal variations in hyporheic zone microbial communities are a key, but understudied, component of riverine biogeochemical function. Here, to investigate the coupling among groundwater-surface water mixing, microbial communities and biogeochemistry, we apply ecological theory, aqueous biogeochemistry, DNA sequencing and ultra-high-resolution organic carbon profiling to field samples collected across times and locations representing a broad range of mixing conditions. Our results indicate that groundwater-surface water mixing in the hyporheic zone stimulates heterotrophic respiration, alters organic carbon composition, causes ecological processes to shift from stochastic to deterministic and is associated with elevated abundances of microbial taxa that may degrade a broad suite of organic compounds.
Quantifying community assembly processes and identifying features that impose them
DOI:10.1038/ismej.2013.93 [本文引用: 1]
Estimating and mapping ecological processes influencing microbial community assembly
DOI:10.3389/fmicb.2015.00370
PMID:25983725
[本文引用: 1]
Ecological community assembly is governed by a combination of (i) selection resulting from among-taxa differences in performance; (ii) dispersal resulting from organismal movement; and (iii) ecological drift resulting from stochastic changes in population sizes. The relative importance and nature of these processes can vary across environments. Selection can be homogeneous or variable, and while dispersal is a rate, we conceptualize extreme dispersal rates as two categories; dispersal limitation results from limited exchange of organisms among communities, and homogenizing dispersal results from high levels of organism exchange. To estimate the influence and spatial variation of each process we extend a recently developed statistical framework, use a simulation model to evaluate the accuracy of the extended framework, and use the framework to examine subsurface microbial communities over two geologic formations. For each subsurface community we estimate the degree to which it is influenced by homogeneous selection, variable selection, dispersal limitation, and homogenizing dispersal. Our analyses revealed that the relative influences of these ecological processes vary substantially across communities even within a geologic formation. We further identify environmental and spatial features associated with each ecological process, which allowed mapping of spatial variation in ecological-process-influences. The resulting maps provide a new lens through which ecological systems can be understood; in the subsurface system investigated here they revealed that the influence of variable selection was associated with the rate at which redox conditions change with subsurface depth.
Resistance, resilience, and recovery of dryland soil bacterial communities across multiple disturbances
DOI:10.3389/fmicb.2021.648455
URL
[本文引用: 1]
Dryland ecosystems are sensitive to perturbations and generally slow to recover post disturbance. The microorganisms residing in dryland soils are especially important as they contribute to soil structure and nutrient cycling. Disturbance can have particularly strong effects on dryland soil structure and function, yet the natural resistance and recovery of the microbial components of dryland soils has not been well documented. In this study, the recovery of surface soil bacterial communities from multiple physical and environmental disturbances is assessed. Samples were collected from three field sites in the vicinity of Moab, UT, United States, 6 to 7 years after physical and climate disturbance manipulations had been terminated, allowing for the assessment of community recovery. Additionally, samples were collected in a transect that included three habitat patches: the canopy zone soils under the dominant shrubs, the interspace soils that are colonized by biological soil crusts, and edge soils at the plot borders. Field site and habitat patch were significant factors structuring the bacterial communities, illustrating that sites and habitats harbored unique soil microbiomes. Across the different sites and disturbance treatments, there was evidence of significant bacterial community recovery, as bacterial biomass and diversity were not significantly different than control plots. There was, however, a small number of 16S rRNA gene amplicon sequence variants that distinguished particular treatments, suggesting that legacy effects of the disturbances still remained. Taken together, these data suggest that dryland bacterial communities may possess a previously unappreciated potential to recover within years of the original disturbance.
Evaluation of vegetation biomass carbon storage in deserts of Central Asia
中亚干旱荒漠区植被碳储量估算
The unseen majority: Soil microbes as drivers of plant diversity and productivity in terrestrial ecosystems
DOI:10.1111/j.1461-0248.2007.01139.x
PMID:18047587
[本文引用: 1]
Microbes are the unseen majority in soil and comprise a large portion of life's genetic diversity. Despite their abundance, the impact of soil microbes on ecosystem processes is still poorly understood. Here we explore the various roles that soil microbes play in terrestrial ecosystems with special emphasis on their contribution to plant productivity and diversity. Soil microbes are important regulators of plant productivity, especially in nutrient poor ecosystems where plant symbionts are responsible for the acquisition of limiting nutrients. Mycorrhizal fungi and nitrogen-fixing bacteria are responsible for c. 5-20% (grassland and savannah) to 80% (temperate and boreal forests) of all nitrogen, and up to 75% of phosphorus, that is acquired by plants annually. Free-living microbes also strongly regulate plant productivity, through the mineralization of, and competition for, nutrients that sustain plant productivity. Soil microbes, including microbial pathogens, are also important regulators of plant community dynamics and plant diversity, determining plant abundance and, in some cases, facilitating invasion by exotic plants. Conservative estimates suggest that c. 20 000 plant species are completely dependent on microbial symbionts for growth and survival pointing to the importance of soil microbes as regulators of plant species richness on Earth. Overall, this review shows that soil microbes must be considered as important drivers of plant diversity and productivity in terrestrial ecosystems.
Conceptual synthesis in community ecology
DOI:10.1086/652373 URL [本文引用: 2]
Bridging rare and abundant bacteria with ecosystem multifunctionality in salinized agricultural soils: From community diversity to environmental adaptation
Responses of different life-form plants in Garbantunggut Desert to small rainfall events
古尔班通古特沙漠不同生活型植物对小雨量降雨的响应
Distinctive pattern and mechanism of precipitation changes affecting soil microbial assemblages in the Eurasian steppe
DOI:10.1016/j.isci.2022.103893 URL [本文引用: 1]
Effects of extreme drought on community and ecological network of soil fungi in a temperate desert
DOI:10.17520/biods.2021327
[本文引用: 3]
<p id="p00005"><strong>Aims</strong> Extreme drought exacerbates the expansion of desert areas around the world. Microbial diversity is associated with multiple ecosystem functions in the desert. Evaluating the response of fungal communities to extreme drought is essential for our understanding of regional desertification caused by drought in a temperate desert.</p><p id="p00010"><strong>Methods</strong> Based on three-year (D3) and ten-year (D10) drought plots established in the Gurbantunggut Desert, we investigated the effect of extreme drought on the diversity and ecological network of fungal communities.</p><p id="p00015"><strong>Results</strong> Our results demonstrated that in both the D3 and D10 plots, the droughts had no significant influence on the Chao1 and Shannon diversity indexes of the whole and abundant fungi, while the rare fungal Shannon diversity index significantly increased. Both extreme drought treatments had a noticeable effect on community composition of whole, abundant and rare fungi, with stronger effect on rare fungi (ANOSIM, <i>R</i> = 0.378-0.595, <i>P</i> < 0.01) than that on the abundant fungi (ANOSIM, <i>R</i> = 0.282-0.555, <i>P</i> < 0.01), suggesting that abundant fungi were more resistant to drought than rare taxa. Moreover, beta-diversity of the whole, abundant, and rare fungi decreased significantly in D3 and D10 treatments, suggesting that extreme drought served as an ecological filter on fungal community assembly. Molecular ecological network analysis revealed that in both the D3 and D10 plots there was a reduced fungal network complexity, suggesting that extreme drought reduced the interactions among fungal communities. In addition, abundant fungi had higher node topological parameters (<i>P</i> < 0.05), indicating that abundant fungi were important for maintaining fungal species interactions under extreme drought conditions.</p><p id="p00020"><strong>Conclusion</strong> Extreme drought significantly altered fungal community composition and weakened the interactions among fungal communities in a temperate desert. Furthermore, rare fungi were sensitive to extreme drought, contributing to reducing the lag in the response to fungal communities, and abundant fungi, as the core microflora in fungal networks, were crucial to sustaining the stability of fungal communities and interactions among species under extreme drought conditions.</p>
极端干旱对温带荒漠土壤真菌群落和生态网络的影响
DOI:10.17520/biods.2021327
[本文引用: 3]
评估极端干旱对温带荒漠土壤真菌群落的影响有助于进一步认识干旱导致的区域荒漠化特征。本研究利用在古尔班通古特沙漠建立的干旱三年和干旱十年样地, 分析了长期极端干旱对温带荒漠土壤真菌群落和生态网络的影响。结果显示, 干旱三年与干旱十年处理对总真菌和丰富真菌的Chao1和Shannon多样性指数均无显著性影响, 而对稀有真菌的Shannon多样性指数有显著促进作用; 干旱三年和干旱十年处理显著影响总真菌、丰富和稀有真菌的群落组成, 且极端干旱对稀有真菌群落变异的影响(ANOSIM, R = 0.378-0.595, P < 0.01)大于对丰富真菌的影响(ANOSIM, R = 0.282-0.555, P < 0.01), 表明丰富真菌具有更强的干旱抵抗力; 另外, 极端干旱显著降低了总真菌、丰富和稀有真菌的β多样性, 表明极端干旱具有生态过滤作用。分子生态网络结果显示, 干旱三年与干旱十年处理降低了荒漠土壤真菌群落网络复杂性, 表明极端干旱减弱了真菌物种间的相互作用; 相比稀有真菌, 丰富真菌具有更高的节点拓扑参数(P < 0.05), 表明丰富真菌对维持极端干旱下的真菌物种间相互作用的重要性。综上所述, 极端干旱显著改变了荒漠表层土壤真菌群落组成, 减弱了真菌物种间的相互作用; 稀有真菌敏感响应极端干旱, 有利于减缓荒漠土壤真菌群落响应的滞后性; 丰富真菌作为网络的核心菌群, 对维持极端干旱下的真菌群落稳定性以及物种间的相互作用很关键。
Precipitation balances deterministic and stochastic processes of bacterial community assembly in grassland soils
DOI:10.1016/j.soilbio.2022.108635 URL [本文引用: 5]
Negative effects of multiple global change factors on soil microbial diversity
DOI:10.1016/j.soilbio.2021.108229 URL [本文引用: 1]
The differentiation of soil bacterial communities along a precipitation and temperature gradient in the eastern Inner Mongolia steppe
DOI:10.1016/j.catena.2017.01.007 URL [本文引用: 2]
Plant species diversity and community classification in the southern Gurbantunggut Desert
DOI:10.5846/stxb URL [本文引用: 1]
古尔班通古特沙漠南部植物多样性及群落分类
Spatial patterns of microbial nitrogen-cycling gene abundances along a precipitation gradient in various temperate grasslands at a regional scale
DOI:10.1016/j.geoderma.2021.115236 URL [本文引用: 1]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}