



家养动物的驯化起源一直是多个学科领域关注的热点方向。对驯化起源的研究有助于我们更深入地了解农业和人类文明历史, 甚至包括人类语言和文化的传播(Diamond & Bellwood, 2003), 为解读现代文明的根源提供视角。
1 驯化起源
1.1 驯化的时间与地点
新石器时代早期, 采集者和狩猎者依靠与野外动植物相处的经验摸索出更好应对气候变化的集约化获取食物的方式, 由此产生了农民和农耕经济。除了狗可能是唯一在农耕经济出现之前被驯化的动物外, 大规模的驯化事件伴随着农业文明的起始与发展, 更倾向发生在温度适宜、灌溉充足和生态资源丰富的地区。独立的动植物驯化事件至少发生在美洲、西南亚、南亚、东亚等11个地区, 而早期的动物驯化主要集中在近东的新月沃土、中国中部和南美的安第斯山脉(Larson & Fuller, 2014; Larson et al, 2014)。由约旦河、幼发拉底河和底格里斯河共同孕育的新月沃土地区, 产生了世界上最早的农耕文明——美索不达米亚文明。距今一万年前的农耕遗址中便出现了豌豆、亚麻、鹰嘴豆、小麦和大麦等谷物(Brown et al, 2009), 同时, 此地驯化的山羊、绵羊、家牛等农业动物传播到各个大洲; 中国的黄河和长江流域也是早期农耕文明起源地之一, 独立驯化了猪、黍、粟、大豆和稻米等主要农业动植物(Larson et al, 2014); 有“南美洲脊梁”之称的安第斯山脉纵贯南美大陆西部, 美洲驼(Lama glama)和羊驼(Vicugna pacos)凭借草食性生态位优势首先被驯化用于肉食、驼役和生产驼毛制品(Wing, 2011)。表1汇总了主要的家养动物的驯化起源信息。
表1 主要家养动物的驯化起源信息。根据Larson和Fuller (2014)、Larson等(2014)和Teletchea (2019)整理。内容包括地理起源、野生祖先、时间起源(前驯化时期和形态学改变时期)和驯化途径。其中前驯化时期是在发生骨骼形态学和性状变化之前出现的特殊集约化管理现象, 是迈向驯化时必不可少的探索阶段。“×”表示此处目前尚未取得可靠证据或者存在较大争议。
Table1
| 学名 Scientific names | 地理起源 Geographic origin | 野生祖先 Wild ancestor | 前驯化时间(距今年限) Pre-domestication time (BP) | 形态学转变(距今年限) Time of morphological changes (BP) | 驯化途径 Pathway to domestication | ||
|---|---|---|---|---|---|---|---|
| 起始 Start | 完成 Finish | 起始 Start | 完成 Finish | ||||
| 山羊 Capra hircus | 西南亚 Southwest Asia | 野山羊 C. aegagrus | 10,500 | 9,750 | 9,750 | 8,000 | 猎食 Prey |
| 绵羊 Ovis aries | 西南亚 Southwest Asia | 亚洲盘羊 O. orientalis | 10,500 | 9,750 | 9,750 | 8,000 | 猎食 Prey |
| 普通牛 Bos taurus | 西南亚 Southwest Asia | 原牛欧亚亚种 B. p. primigenius | 10,500 | 10,250 | 10,250 | 8,000 | 猎食 Prey |
| 瘤牛 Bos indicus | 南亚 South Asia | 原牛印度亚种 B. p. mauretanicus | × | × | 8,000 | 6,500 | 猎食 Prey |
| 猪 Sus domesticus | 西南亚 Southwest Asia | 欧亚野猪 S. scrofa | 11,500 | 9,750 | 10,250 | 9,000 | 共生 Commensal |
| 东亚 East Asia | 欧亚野猪 S. scrofa | × | × | 8,500 | 6,000 | 共生 Commensal | |
| 美洲驼 Lama glama | 南美洲 South America | 原驼 L. guanicoe | × | × | 6,000 | 4,000 | 猎食 Prey |
| 羊驼 Vicugna pacos | 南美洲 South America | 原驼 L. guanicoe | × | × | 5,000 | 3,000 | 猎食 Prey |
| 马 Equus caballus | 中亚 Central Asia | 野马 E. ferus | 6,750 | 5,500 | 5,500 | 4,000 | 定向 Directed |
| 驴 Equus asinus | 北非 North Africa | 非洲野驴 E. africanus | × | × | 5,500 | 3,500 | 定向 Directed |
| 水牛 Bubalus bubalis | 南亚 South Asia | 野水牛 B. arnee | × | × | 4,500 | × | 猎食 Prey |
| 双峰驼 Camelus bactrianus | 中亚 Central Asia | × | × | × | 4,500 | × | 定向 Directed |
| 单峰驼 Camelus dromedarius | 阿拉伯半岛 Arabia | × | × | × | 3,000 | × | 定向 Directed |
| 鸡 Gallus gallus domesticus | 东亚/东南亚 East Asia/Southeast Asia | 红原鸡 G. gallus | × | × | 4,000 | × | 共生 Commensal |
| 家鸭 Anas platyrhynchos domesticus | 东亚/东南亚 East Asia/Southeast Asia | 绿头鸭 A. platyrhynchos | × | × | 1,000 | × | 共生 Commensal |
然而, 环境是否适合种植与养殖并不是动物驯化发生的绝对条件, 越来越多的研究表明, 能否被成功驯化在于动物本身的特性(Diamond, 2002)。适合驯养的动物具有6项特征: 集群的社会结构; 较易满足的食性(食草性或杂食性); 繁殖能力强; 生长周期短, 生长速度快; 性格温顺, 低侵略性; 对人的忍受力高, 不易逃跑(McCall & Diamond, 1997; Driscoll et al, 2009)。欧亚大陆散布着五大驯化哺乳动物: 牛、绵羊、山羊、猪和马, 而近东地区的瞪羚(Gazella)和非洲的斑马等大型哺乳动物不满足驯化的6项特征至今未能驯化成功(Diamond, 2002)。总体来看, 驯化的持续性、复杂性以及人类的地域文化差异等因素导致成功驯化的事件并不多。不同地区的自然环境、人文环境和发展水平以及动物自身特性等因素综合决定了驯化事件的发生与发展(Losey, 2021), 野生动物最有可能从少数的几个农耕文明中心被成功驯化, 之后传播到世界各地。
1.2 驯化途径和驱动因
除了以人对动物的喜爱出发而逐渐对动物实现繁殖控制的渐进式过程外, 现今广为接受的是共生、猎食和定向3种驯化途径(Zeder, 2012) (表1)。在共生途径中, 野生动物被食物残渣、污秽物吸引, 主动接近人类生态位(human niche)并逐渐与人类建立了双向的伙伴关系。在这一过程中, 人们从无意识到逐步提高驯化的目的性, 例如最早被驯化的狗。捕猎途径初期, 人类会直接在居住地杀死捕获的野生动物, 而后为了最大程度地利用猎物, 人们选择相对温顺的动物进行狩猎和畜群管理, 最终演变为定向繁殖, 例如猪的驯化。定向途径在人们已经掌握了一定驯化经验之后才会发生, 伴随着人们明确的驯化目的。该途径所需要的驯化时间相对较短, 例如马、骆驼、牛等的驯化(Larson & Burger, 2013; Larson & Fuller, 2014)。
一些研究者从更细致的角度来研究动物驯化的驱动因, 认为驯化受到一系列复杂的、以当地生态和文化为依据的多种因素影响(Zeder & Smith, 2009), 和不同地区的驯化策略与农业发展程度相关。袁靖和董宁宁(2018)从社会内部的动因、人与动物的关系、文化生态学的角度全面地总结了国内外学者提出的驱动因, 包括夸富宴、祭祀说、宠物饲养、最佳觅食理论、文化生态位构建、肉食说等。以3种驯化途径之一的猎食途径为例来看, 驯化的驱动因素可以是多种多样的。人们追求觅食效率, 习惯于得到饮食结构中能量回报比高的食物, 进而对周边的小型野生动物展开了驯化, 即最佳觅食理论(Optimal Foraging Theory, OFT)中的饮食宽度模型(Diet Breadth Model, DBM) (Zeder, 2015)。然而有些情况下, 驯化可能是被动的。当周边可获取的野生资源不足以满足自身的需求时, 例如渔猎和采集经济方式难以应付饥荒、存储的猎物短缺等情况下发生的驯化事件, 即符合被动发展理论和肉食说(罗运兵和李想生, 2012)。人们还可以自发地修饰、整合所处的自然环境, 成为自身和其他生物在选择进化中的引导者, 即文化生态位构建理论(Cultural Niche Construction, CNC) (Laland et al, 2010)。
总之, 驱动因素是相当复杂的, 并且多种驱动因素可能同时存在, 即相同的物种和地区中存在不同的驯化驱动因素。人们目前依靠考古遗址的群体结构、文化风俗、社会形态等来综合推测当地动物驯化的原始目的。
1.3 驯化后的扩散及品种选育
2 研究家养动物驯化起源的材料和方法
2.1 考古学方法
家养动物通常在形态特征上与野生祖先表现出差异。吻部缩短、面部凹陷、脑容量减少和齿列扭曲等形态特征常常综合起来用作判断家猪和家狗驯化的依据(Cucchi et al, 2011)。然而这些形态变化不一定同时发生且需要大量的时间累积, 不能准确地追溯驯化早期阶段(Arbuckle, 2005)。新兴的臼齿几何形态学测量(Geometric Morphometrics, GM)在近几年非常受欢迎。通过GM提取样本特征并对应到二维或三维空间的计量点, 结合所有点坐标推断出样本形状, 不仅可以更准确地量化样本形状和比例关系, 也有助于复杂结构的可视化(Zelditch et al, 2004)。Cucchia等(2011)将现代样本和贾湖遗址出土的猪化石的M3臼齿进行几何形态学分析和普氏分析, 推测最晚在距今8,500年的贾湖二期遗址已经存在家猪, 并将贾湖遗址重新确定为中国最早的独立驯化中心。此外, 颅骨高度和长度的比例、鼻子高度与颅骨总长度的比例也被用作区分狼头骨和狗头骨(Pitulko & Kasparov, 2017)。
家养动物饮食与生活习惯的改变, 以及过度劳役可能造成一些病理现象。线性牙釉质发育不全(Linear Enamel Hypoplasia, LEH)是一种基于发育障碍的牙齿疾病, 不仅可以用于研究家养动物由于环境变化导致的生理紧张, 也广泛用于研究尼安德特人、新石器时代古人、黑猩猩的早期生态压力和适应性扩张(Guatelli-Steinberg et al, 2004; Guatelli- Steinberg et al, 2012; Orellana-González et al, 2020)。营养不良、维生素缺乏或疾病干扰了成釉细胞的釉质分泌阶段, 釉质不足则导致发育后的齿冠表面出现凹痕或横线(Orellana-González et al, 2020)。然而, 由于动物生长的环境压力较为复杂, 具有较高的LEH发病率不能作为判断家养动物的充分条件。例如, 家猪除了会因为断奶或者第一次过冬产生M1、M2臼齿LEH外, 还可能因为人们的狩猎刺激在M3臼齿留下病理学痕迹(Katzenberg, 2008)。除了LEH, 因为劳役动物产生的骨质增生、摄入高淀粉和高糖食物引发的齿槽脓肿等病理学现象都可通过考古发现(袁靖, 2015)。不同的病理学现象可能提示了不同的驯化方式。
骨胶原中的碳氮稳定同位素分析是广泛使用的动物食性判定方法, 可识别动物在不同时期的膳食特征及变化(Katzenberg, 2008)。由于不同植物依赖于不同的光合作用途径(C3或C4), 碳氮稳定同位素可以反映动物的饮食来源。稳定的氮同位素值会随着食物链中营养级的上升而增加(每个营养级在3%和5%之间), 因此依据δ15N可以区分食草、杂食或肉食性动物(Guiry & Grimes, 2013)。家养动物一般以人类的剩饭、农作物副产品为食, 因此考古学将人与动物骨骼的碳氮稳定同位素的相关性作为判断家养动物的依据。这种方法同时也为研究史前人类饮食结构提供了宝贵的证据。研究发现, 阿拉斯加西部沿海地区的狗骨胶原中δ13C和δ15N高于正常哺乳动物, 推测与当地居民一样主要以鲑鱼为食(Guiry & Grimes, 2013), 可以作为已驯化的证据。
通常, 完整遗址中会出现多种动物的遗骸。统计分析同种群体占整个遗址中所有动物的比例以及该群体内部的年龄结构和性别特征等手段可用来了解人类早期的动物管理模式, 进而推断是否发生驯化。以肉食为驱动因的家养动物一般在遗址中占有较高的比例。为了更优的投入回报比, 人们屠宰幼年的雄性个体, 保留雌性, 以便高效地可持续化饲养, 这种有意识的管理模式的出现早于动物形态学上的变化。在距今约10,000年的扎格罗斯山脉遗址中, 发现了大量被宰杀的幼年雄性家羊, 这些个体比形态学上发生体型变化和羊角形状变化的家羊早500‒1,000年(Zeder, 2008)。但是, 特殊的管理模式不一定代表发生驯化, 它也可能出现在随机的捕猎活动中或发生在前驯化时期(proto-domestication) (Rowley-Conwy et al, 2012)。例如在越南北部巴曼克遗址中, 有64%的猪在12个月大之前被宰杀, 只有9%的猪存活超过4年, 但是基于磨牙数据的测量分析显示这些个体与当地野猪没有显著差异(Jones et al, 2019), 仅能表明此时出现了特殊的管理方式。
目前, 考古学方法推断家养动物的驯化起源是比较令人信服的方式, 但没有哪一种具体方法是完全准确且全面的。由于处于驯化早期的动物形态变化在考古学上难于区分, 大大影响了驯化起始时间的估计。考古人员一般搜寻遗址中观察到的全部可用信息, 综合各项指标判断是否为家养动物。若该地为独立的驯化中心, 则形态和结构特征会随着遗址的地层时间发展显示出历时性变化, 从而反映了当地人从控制管理野生动物到逐渐习得成熟的驯化技术的过程。
考古学是一门不断发展、不断进步的学科。随着科技的发展, 考古学结合数理统计、现代分子遗传学、古基因组学等方法, 为传统的考古注入了新的力量。
2.2 分子遗传学方法
2.2.1 分子材料
驯化研究常用的分子材料是线粒体DNA (mtDNA)和核基因组DNA (nuclear DNA), 来源于现代样本和古DNA。高通量测序技术使得高覆盖度和高测序深度的现代样本基因组信息更容易获得, 从化石中提取的古DNA在时间深度上也为研究提供了非常重要的细节信息。高分辨率的基因组信息配合群体遗传学手段可以准确地推断驯化事件发生的时间, 重建复杂的驯化历史。
(1)线粒体DNA和核DNA。线粒体DNA是进化遗传学领域常用的分子材料。线粒体DNA具有拷贝多、易于获取、D-loop区进化速度快、成本低等优势, 常用于构建物种之间的系统发育关系以及研究系统地理学和群体统计学特征(Orlando & Cooper, 2014)。但是, 线粒体DNA是非重组的单亲遗传标记, 包含的绝对信息量少, 缺乏精确定量种群混杂程度的能力, 可能会造成系统误差和推论的片面性(Larson & Burger, 2013)。例如, 线粒体系统发生树显示尼安德特人和现代人是没有杂交信号的姊妹群体, 而在核基因组上观察到二者具有一定比例的混合(Sánchez-Quinto & Lalueza-Fox, 2015)。另外, 瓶颈效应通常影响了遗传多样性。而线粒体数据相较于核基因组来说, 对过去发生的中度和轻度的瓶颈事件并不敏感(Mourier et al, 2012)。
核DNA指真核生物细胞核染色体的遗传信息。进化中的自然和人工选择、DNA重组、基因交流和群体大小变化等事件, 都会在核DNA中留下痕迹, 且不会像线粒体DNA那样丢失父系的信息, 是更丰富和全面的研究对象。十几年来蓬勃发展的下一代测序技术(Next Generation Sequencing, NGS)解决了全基因组测序数据量大、耗时长和费用高等难题, 使得单碱基测序成本、测序片段长度、总体测序量和能获取的基因组变异信息都有了大幅改善(Goodwin et al, 2016), 推动了对核基因组的充分利用。全基因组单核苷酸多态性位点(Single Nucleotide Polymorphism, SNP)是单碱基位点发生的等位基因变异, 在人类基因组变异信息中占比约90% (Varela & Amos, 2010), 是目前遗传分析中使用最广泛的标记。
(2)古DNA。现代样本的遗传数据能用于回溯祖先驯化历史, 以此推断种群遗传结构、驯化中心及可能的驯化起始时间。但是, 仅基于对现代样本的分析来推断驯化历史是否足够有效还存在争议(Frantz et al, 2020)。古DNA能在时间跨度上提供更为直接的证据, 并反映家养动物驯化的初始状态。古DNA最初非常难于获得, 在成千上万年的遗留中易发生交联、脱氨、碎片化而渐渐失去有效信息(Allentoft et al, 2012), 其中核基因组衰变速度至少是线粒体基因组的两倍(Willerslev et al, 2003)。内源古DNA还会被土壤环境中的微生物DNA严重污染, 即便DNA提取成功后可信度也大打折扣。
解决上述问题的重大突破是测序技术中的古DNA捕获技术。它利用特定的寡核苷酸分子探针与构建的DNA总文库杂交得到内源靶序列, 从本质上提升了古DNA提取的效率(Maricic et al, 2010; Haak et al, 2015)。Fu等(2013)通过该方法得到了距今约4万年北京田园洞人的mtDNA (35.6-flod)和21号染色体的核DNA数据, 成为揭示东亚人遗传起源的重要样本。DNA捕获技术结合聚合酶链反应(PCR)、高通量测序技术, 使得丰度低、碎片化程度很高的古基因组DNA可以有效地被富集、扩增, 并建库进行高通量测序, 重现内源分子的遗传信息(Pääbo et al, 1989; Gansauge & Meyer, 2013; MacHugh et al, 2017)。此外, 考古学家开创了从毛发和骨骼密集的硬组织(牙齿和颞骨)中提取古DNA的方法, 进一步显著提升了内源性古DNA的产量(Hänni et al, 1990; Der Sarkissian et al, 2015; Pinhasi et al, 2015)。
自2013年第一个野马古基因组被发表后, 家养动物及其野生祖先的古基因组发表数量迅速增加, 丰富了我们对于驯化初始过程的理解(Orlando et al, 2013; Frantz et al, 2020)。之前, 人们认为家养动物在初始驯化后和其野生种群会产生生殖隔离, 而众多家养动物的古DNA研究表明, 野生种群对家养动物存在普遍且持续的基因渗入(Frantz et al, 2015, 2020)。历时性的古DNA更深入和全面地揭示了整个驯化起源和扩散地时空地理框架。旧石器时期至中世纪的山羊古DNA样本表明, 早期家羊在近东西部、东部和西南部存在不同的野山羊遗传背景, 提示了多重平行的驯化起源的可能性(Daly et al, 2018)。古DNA还表明, 扩散后的家养动物经当地野生种群基因组的不断渗入而改变了原本的遗传结构, 有时甚至会发生种群替换现象, 而基于现代基因组的地理谱系分析无法揭示这样的信息。
古DNA是很好地了解种群进化、迁移和混杂事件的分子材料, 然而可使用的古代样本是非常有限的。古DNA容易被污染和降解, 因此在温暖潮湿的热带地区, 家养动物的种群历史难以用古DNA来追踪; 即便在适宜的环境中, 由于几千年来人类活动的影响, 发掘的高质量遗骸也是屈指可数的, 很难达到全时空尺度和群体遗传学的数量要求; 家养动物的野生祖先、过渡形态尚不能完全确定, 也会对依赖古DNA的研究结果的真实性造成困扰; 此外, 一般认为化石仅能提供驯化时间估计的下限。因此通过古DNA的方法推断家养动物的驯化起源仍然具有挑战性。
2.2.2 策略与方法
驯化在基因组上留下了可追溯的痕迹, 分子遗传学分析基于种群间、种群和祖先之间的遗传相似性和遗传多态性差异这两种策略推断驯化起源并重建驯化历史(陈善元和张亚平, 2006)。另外, 普遍的基因交流事件也改变了种群间的遗传结构, 使得驯化历史更为复杂, 因此越来越多的学者采用基因渗入工具丰富驯化历史的推断。研究人员基于这些策略基础开发了许多遗传统计量和统计模型。
(1)遗传相似性。研究者们通过构建DNA系统发育树获得拓扑分支结构(Larson et al, 2005; Larson et al, 2010; Kang et al, 2016), 再依据先验信息(已知进化关系、地理关联、时间对应信息)确定进化树的根信息, 从而判断家养动物的亲缘关系和谱系演变。这种方法正是基于样本间序列相似性的思想——家养种群与其祖先种群之间的遗传相似性更高, 共享了更多的变异位点和单倍型种类(图1)。在分子钟理论下, DNA序列以近似恒定的速率发生变异, 因此一个进化分支上所累积的变异数目与该分支的独立分化时间成正比(Ho & Duchene, 2014)。经由化石校准后的系统发生树也用于推断物种或品种的分歧时间。自大量研究转向全基因组SNP位点后, 新开发了许多祖源推断方法。血缘同一性(identity by descent, IBD)是不同个体从共同祖先遗传而来的共享基因片段, 指示了个体间的亲缘关系。一般来说, 亲缘关系近的个体IBD概率高且共享更长的相似性序列片段(Browning & Browning, 2012)。
图1
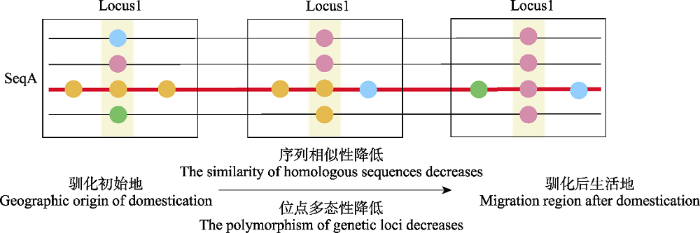
图1
基于分子手段判断驯化起源的策略示意图。不同颜色的圆圈代表不同的基因型。纵向为基因座, 指示位点多态性; 横向将基因座连成序列, 指示序列相似性。伴随驯化的过程, 动物从驯化初始地迁移到各地, 同源序列的相似性降低, 迁移样本在同源位点上的多态性降低。
Fig. 1
Strategies for inferring the origin of domestication based on molecular evidences. Circles with different colors represent different genotypes. The vertical direction represents allelic polymorphism of each genetic locus; the horizontal sequence represents the nucleotide sequence. During the process of domestication, animals migrate from domestication site to other places, with decreased similarity in nucleotide sequences and decreased polymorphism of genetic loci.
全局祖源分析和局部祖源分析是近年来发展较快的基于序列相似性的祖源推断方法。全局祖源分析用于估计混杂个体中不同祖先成分的遗传比例, 有基于降维和聚类、线性回归、基因频率混合分布和概率模型等策略。基于概率模型的全局祖源分析是目前使用最普遍的方法, 常用工具有STRUCTURE和ADMIXTURE, 它们基于贝叶斯算法、马尔科夫蒙特卡洛(Markov Chain Monte Carlo, MCMC)抽样法或最大似然估计, 依据设定的祖先个数K和群体的等位基因频率对观测个体的等位基因建模, 把全局祖源当作模型参数进行估计(Pritchard et al, 2000; Alexander et al, 2009)。局部祖源分析的目的并不是估计个体或群体基因组整体的混合比例, 而是将全基因组拆分成小窗口, 通过贝叶斯、随机森林等分类器来估计每个窗口的祖先来源, 然后采用动态规划、隐马尔可夫及其扩展模型对连续窗口进行统计建模, 估计各基因组区段的祖先来源(Wu et al, 2021), 目前常用的软件有RFMIX、HAPMIX、LAMP-LD等(Wang et al, 2020a)。全局和局部祖源推断基于遗传相似性思想, 与构建系统发生树时需要提供进化根一样, 也需要提供候选的祖先群体来指定遗传混杂发生的方向。
(2)遗传多态性。除了以遗传相似性为出发点以外, 基于遗传多态性来判断家养动物的起源也是一个常用的策略。该策略认为驯化符合奠基者效应——小部分个体从野生种群分离迁出变成新的驯化群体, 因此越接近野生祖先的种群的有效群体大小和多态性越高(图1)。通过计算简单的统计量如杂合度、群体分化指数(Fst)等, 便可以大致推断种群分离路径和群体迁移历史。
杂合度评价基因座上不同等位基因的比例, 是种群遗传多态水平的常用统计量。一般认为驯化起源地的杂合度最高, 在没有频繁基因交流的情况下, 驯化群体的杂合度由于奠基者效应、选择效应和遗传漂变而持续降低(Li et al, 2008)。除了评价单群体的杂合度水平以外, 研究者也通常将观察到的两群体间加权平均杂合度与哈迪-温伯格平衡下的预期杂合度作比较, 衡量群体之间的分化程度, 即Fst。研究群体与野生群体的分化程度低提示该群体处于驯化起始阶段, 与野生种群的分化程度高则提示其处于定向选择阶段或拥有长期的驯化历史。家犬的驯化起源地争议很大, 而研究表明东亚土狗拥有较高的mtDNA、Y染色体遗传多态性, 再结合形态学相似性和群体动态历史等证据, 推测家犬更有可能驯化于东亚南部(Brown et al, 2011; Wang et al, 2016)。
需要指出的是, 越来越多的学者认为遗传多态性用于判断驯化起源可能存在一些问题。近些年的驯化研究表明, 驯化物种遗传多态性的下降可能更多是由于近几百年的品种培育, 而非由于驯化初始阶段(Wang et al, 2014)。并且, 家养动物在初始驯化过程经历的奠基者效应比我们想象得更温和, 加之随后与野生种群普遍发生基因交流事件, 导致现代部分家畜相比其野生祖先观察不到多态性丢失(Wang et al, 2014; Bosse et al, 2019; Wang et al, 2021), 相反野生种群可能由于过度捕杀而丢失多态性。此外, 有些家养动物的野生祖先已经消失, 使得利用与野生型进行多态性比较的策略失效, 例如家牛的祖先——曾经遍布欧亚大陆的原牛已彻底灭绝(Bos primigenius) (Park et al, 2015)。
(3)驯化中的基因交流。研究发现, 动物驯化的过程不是链条式, 而是呈现一种网状结构, 即在驯化初始阶段和驯化过程中伴随着大量的基因交流事件(Frantz et al, 2015)。近年来不断开发的群体遗传学分析方法擅长在群体基因组中寻找驯化中的基因交流信号, 常用的有f统计量和D统计量。
f统计量(f-statistics)包括三群体f3、四群体f4统计量。f3 (A, B; C)、f4 (A, B; C, D)计算种群之间等位基因频率差的乘积, 即: (c‒a)(c‒b)和(a‒b)(c‒d), 依据统计量的正负来判定渗入方向及信号强弱(Reich et al, 2009; Patterson et al, 2012)。若f3统计量为负, 提示群体C由群体A和群体B混杂而来。f4统计量在零假设时(群体间没有混杂)期望值为0, 如果f4的观测值为正, 则提示AC或BD群体之间存在混杂, 如果f4的观测值为负, 则提示AD或BC群体之间存在混杂(文子龙和赵毅强, 2021)。outgroup f3是f3的一种特殊情况, C代表种群A与B的外群而非A与B的混杂后代, 该统计量实际上是估算外群C到种群A与B的共同祖先的分支长度。Admixtools中的qpAdm、qpWave程序可以基于F统计量结果得到众多群体间的混合事件、混合权重和分化支, 最终由qpGraph程序拟合成完整的混合分支图(Patterson et al, 2012)。D (H1, H2, H3, H4)同样使用4个群体进行基因渗入检验。其中H4为外群, 基于ABBA-BABA模型计算两个姊妹群体H1, H2与渗入源群体H3之间的共享等位基因数目的差异, 若D统计量小于0, 则H1与H3发生过基因交流, 反之则H2与H3发生过基因交流(Soraggi et al, 2018)。
2.3 比较与讨论
考古学与分子遗传学判断家养动物驯化起源的策略多种多样, 由于每项研究的数据类别、数据量和分析重点不尽相同, 单用某种策略会导致结果不具有全面性, 甚至存在较多冲突。因此实际研究中考虑整合多种研究策略, 尽可能完整地描绘出家养动物的驯化历史。例如, 遗址中遗骸的年龄结构、性别结构、饮食结构分析的证据可能仅指示了前驯化时期的特殊管理方式, 并不能确定驯化事件的发生; 遗传多态性策略本身便具有一定缺陷, 需要其他策略辅助验证其推论的真实性; 近些年发展较快的祖源分析、种群动态历史分析等方法虽然可以提供驯化中多个事件发生的分子证据, 但是不足以推断驯化发生的完整地理框架。如果分子水平的结果能从不同历史时期和地点的古DNA数据中获得验证, 结论会更加可信。总之, 家养动物的驯化起源和历史是极其复杂的, 需要不同角度的方法与策略联合推断才能得到较为全面的结论。
3 主要家养动物驯化起源研究进展
家猪拥有约10,000年的驯化历史, 在中国有明确的独立驯化的证据(Ervynck et al, 2001; Cucchi et al, 2011)。中国拥有约100余个家猪品种, 占全球总资源的1/3 (Ai et al, 2015)。考古学证据表明, 家鸡驯化于至少距今4,500-4,000年左右(Larson & Fuller, 2014), 中国是其可能的驯化地之一。家鸡是表型和用途最为丰富的驯化动物, 为人类提供优质低价的蛋白来源, 也用于观赏消遣。经济合作与发展组织 (OECD)和联合国粮食及农业组织(FAO)预测2025年我国猪肉消费规模将达到6,510‒10,010万吨, 占所有肉类消费的2/3以上, 禽类消费量紧跟其后, 并且会成为在世界范围内唯一超越猪的肉食来源(朱文博和陈永福, 2018)。山羊、绵羊、家马以及家牛也是早期驯化的重要家畜, 对人类文明和社会发展具有重要意义。其中山羊、绵羊和家牛都是由西南亚新月沃土区域驯化后传播到欧洲和中国, 拥有相似的驯化时间和传播路径, 而家马的驯化起源目前还没有定论。
3.1 家猪
现代家猪起源于欧亚野猪(Sus scrofa) (Frantz et al, 2016)。欧亚野猪在距今约400万年的上新世从东南亚岛屿迁移到欧亚大陆上。由于拥有高纬度和不同环境温度的高适应性, 欧亚野猪迅速替代了侏儒猪(Porcula)以外的欧亚大陆上的其他猪种(Frantz et al, 2016)。欧亚野猪在100万年前分化成欧洲野猪和亚洲野猪两支(Bosse, 2018), 各自独立驯化, 是当今唯一被驯化成家猪的猪科动物。多名学者提出, 猪科动物在更新世可能存在大量的种间基因交流。例如在X染色体上的一段非重组区域, 中国北方野猪与欧洲野猪聚在一簇, 而中国南方野猪却与东南亚岛屿的野猪聚在一起, 推测欧亚野猪在进入欧亚大陆时可能与现已灭亡的其他欧亚种群发生了适应性渗入事件(Ai et al, 2015)。另外, 欧洲和中国北方野猪在卡拉布里亚阶至末次盛冰期的冰期间隙可能存在长期的基因流动(Groenen et al, 2012)。这些事件进一步提升了欧亚野猪的适应力, 使得它们迅速扩张到欧亚大陆各处(Liu et al, 2019)。
目前考古学证据提示家猪至少在两个地方独立驯化: 近东的小亚细亚半岛和中国。距今约9,000年前(地层年龄为12,000至8,300年)的土耳其东部Çayönü Tepesi、Hallan Çemi Tepesi和Tell Hallula等遗址发现了介于家养和野生种群的过渡态猪遗骸(图2C), 它们死亡年龄较小、M3臼齿变短、线性牙釉质发育不全且呈现历时性趋势(Mason, 1984; Ervynck et al, 2001; Frantz et al, 2016)。这是迄今为止世界范围内比较明确的、较早出现家猪的考古证据。中国家猪的化石首先发现于黄河流域的河南舞阳贾湖遗址(图2A), 基于臼齿几何结构、齿列扭曲、肉量比例、猪下颌骨陪葬等多个典型特征判断在距今8,500年前已经存在家猪(罗运兵和张居中, 2008; Cucchi et al, 2011)。同近东一样, 中国家猪饲养现象不局限于单一的位置。考古学证据提示, 浙江跨湖桥遗址、河北武安磁山遗址也可能是新石器时代独立的家猪驯化地点(Jing & Flad, 2002; Frantz et al, 2016) (图2A), 其中处于中国南方地区跨湖桥遗址出土的家猪体型较小, 这种体型差异恰好对应了中国南北方野猪的体型差异(袁靖, 2015), 并提示中国境内家猪可能的多起源。
图2
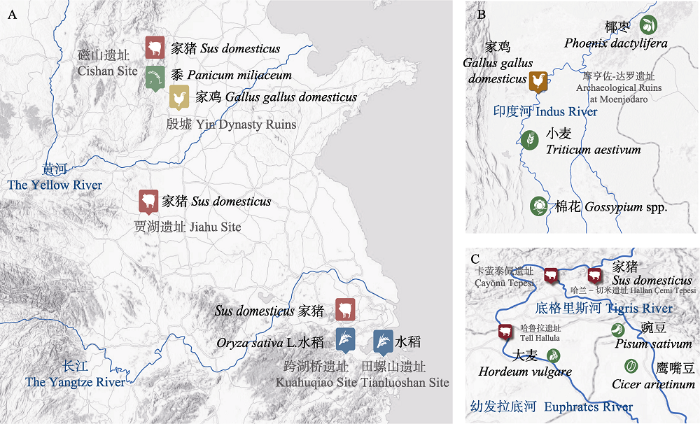
图2
家猪与家鸡早期考古遗址及周边河流、农作物驯化情况。(A)中国长江黄河孕育的中华文明。(B)印度河孕育的印度河文明。(C)新月沃土孕育的美索不达米亚文明。
Fig. 2
Early archaeological sites of domesticated pigs and chickens, with surrounding rivers and crops. (A) Chinese civilization nurtured by the Yangtze River and Yellow River. (B) The Indus civilization nurtured by the Indus. (C) Mesopotamian civilization nurtured by the Fertile Crescent.
然而, 许多基于线粒体和核基因组系统发生学的研究提出了不同的看法。在中国, 除去黄河中游流域的古代家猪与现代家猪具有连续的共享单倍型, 是较公认的驯化中心以外, 湄公河流域、长江中下游流域、青藏高原等地在时空谱系和主成分分析上都存在独立驯化的迹象(Wu et al, 2007; Tong et al, 2020)。在世界范围内, 有学者提出除近东和中国大陆之外, 印度、欧洲、东南亚岛屿、泰国、缅甸也可能是家猪独立的驯化中心(Larson et al, 2005)。但是, 遗传谱系上与当地野猪相似、与其他地域品种的血统分离并不意味着在当地发生了独立驯化(Larson & Burger, 2013)。古DNA分析表明, 携带着mt-Y1近东线粒体单倍型的家猪于8,000年前随着近东农民扩散至欧洲, 而3,000年后无论是线粒体还是核基因组, 近东血统完全被欧洲当地的野猪替换(Frantz et al, 2019)。这种本地野生种群替代迁入种群的现象与二者生殖隔离小、最初迁入种群较小以及欧洲松散的家猪管理方式有关(Porter, 1993; Currat et al, 2008)。同样地, 东南亚岛屿作为现代野猪的起源地、大洋洲家猪的遗传来源地, 这里的家猪形成独立的分支但是缺少考古学证据, 也可能是外地迁徙来的家猪与当地野猪不断杂交导致了遗传成分发生替换(Larson et al, 2007; Larson & Burger, 2013)。
3.2 家鸡
目前人们普遍认为家鸡的祖先是红原鸡(G. gallus) (Fumihito et al, 1994)。红原鸡在东亚广泛分布, 从巴基斯坦到中国云南、缅甸、印度等大部分地区, 以及苏门答腊岛、爪哇和巴厘岛均有分布(Groeneveld et al, 2010)。红原鸡现存5个亚种: 原鸡指名亚种(G. g. gallus)、原鸡滇南亚种(G. g. spadiceus)、原鸡海南亚种(G. g. jabouillei)、原鸡印度亚种(G. g. murghi)和原鸡印尼亚种(G. g. bankiva), 家鸡最有可能由原鸡滇南亚种驯化而来(Wang et al, 2020b)。近些年关于家鸡皮肤颜色的研究证实了灰原鸡的遗传贡献, 这可能是家鸡驯化之后人为介导的种间交流导致(Eriksson et al, 2008)。
同家猪一样, 基于线粒体和核基因组的研究也提出了家鸡多起源的观点。Fumihito等(1996)首先基于不完整采样的研究提出家鸡可能驯化于泰国(红原鸡栖息地之一)。张亚平等的研究团队基于6个线粒体DNA进化分支的地理分布提出, 家鸡有多个母系地理起源, 包括云南、华南、西南及周边地区, 以及印度次大陆(Liu et al, 2006)。张亚平等人后续的研究发现单倍群E2和E3主要出现在南亚家鸡和红色原鸡中, 单倍群I分布在印度东北部, 单倍群H来源于中国西南地区, 提出红原鸡在南亚、中国西南地区和东南亚地区发生了多次独立驯化的观点(Miao et al, 2013)。最近基于f3、f4、种群动态历史及分子系统发生学的研究发现, 中国西南、泰国、缅甸的本土品种——原鸡滇南亚种在进化树上更靠近现代家鸡, 最有可能是现代家鸡的驯化祖先, 且家鸡在南亚和东南亚扩散时与当地的其他野生亚种发生了基因交流(Wang et al, 2020b)。
新石器时期的中国北方是否独立驯化了家鸡一直是学者们争论的话题。20世纪80年代末有学者提出, 距今8,000年左右的中国北方磁山遗址出土了比原鸡更大的家鸡骨头(West & Zhou, 1988)。2014年赵兴波课题组将中国河北省南庄头遗址、磁山遗址、山东省王因遗址与湖北省九连墩遗址出土的鸡骨提取线粒体DNA后进行序列比对(Xiang et al, 2014), 认为上述中国北方的新石器早期遗址均为家鸡驯化起源地, 并推断家鸡在中国北部、南亚与东南亚多地独立驯化。但是, 该研究有诸多细节存在漏洞, 被多名学者提出质疑(Peters et al, 2015; 袁靖等, 2015): 首先, 文中提到的北方遗址出土的“鸡骨”在种属鉴定上要么论证不清晰, 要么直接把雉误判成了家鸡; 其次, 距今约8,000‒10,000年的河北气候较为干冷, 不适宜适应了温暖湿润环境的红原鸡生存。
鸡作为非常重要的家禽, 目前报道的驯化时间明显晚于狗、猪、牛、马。一是因为祖先红原鸡生活在热带森林和次生竹林的温热气候下, 与生俱来的环境适应性较为局限。二是家养动物驯化一般发生在农耕经济社会形成的前期, 红原鸡的栖息地与农耕文明的起源地(如中国的长江与黄河流域以及新月沃土的两河流域)距离遥远, 因此不是优先被驯化的对象。此外, 相较于其他家养动物化石资源来说, 稀少的鸡化石材料也阻碍了对家鸡驯化时间和地理框架的探究, 使得目前的考古证据和分子证据存在较大差异。
3.3 其他主要家养动物驯化历史
绵羊的野生祖先亚洲盘羊(Ovis orientalis)和山羊的祖先野山羊(Capra aegagrus)于晚中新世时期分化, 并在距今10,500年时在近东的土耳其安纳托利亚和伊朗扎格罗斯分别完成驯化(Alberto et al, 2018)。考古证据表明绵羊的驯化初始于公元前9,000年, Çayönü、Höyük和Nevali Çori遗址首先出现了围畜栏、屠宰幼年雄性个体的集约化羊群管理方式和骨骼形态学转变, 并很快传播到整个新月沃土以及临近地区(Yurtman et al, 2021)。绵羊驯化之初是为了满足人们肉食与奶制品的需要, 而内源性逆转录病毒(enJSRV)与考古学证据都表明, 近东人在公元前4,000年的青铜器时期开始转向对羊毛性状的人工选育, 由此引发了改良后的绵羊的第二次世界范围内的广泛传播(Chessa et al, 2009; Sabatini et al, 2019; Yurtman et al, 2021)。李孟华团队整合分子手段与考古学记录重建了绵羊向中国的传播路径。绵羊与5,000‒5,700年前从近东经过高加索和中亚传播到第二个扩散中心——蒙古高原, 后随着氐羌民族的两次向南扩张逐步传播到黄河中上游、西藏高原、贵州高原(Lv et al, 2015; Zhao et al, 2017)。山羊与绵羊驯化的考古学证据以及向各大洲传播的路线都很相似。古DNA和现代核基因组提示家山羊可能在近东的不同位置经历过多次独立的驯化, 而目前家山羊普遍携带的是起源于安纳托利亚东南部的A型线粒体单倍型(蔡大伟等, 2021)。学者们发现免疫基因MUC6、毛色基因KITLG与KIT、绒毛性状相关基因FGF5在家山羊传播扩散的过程中受到了强烈的选择并固定下来, 为探索山羊驯化后的环境适应性提供了新的线索(郑竹清, 2019; 蔡大伟等, 2021)。
家牛分为普通牛(Bos taurus)和有肩峰的瘤牛(B. indicus), 分别由已经分化至少147,000年的原牛欧亚亚种(B. p. primigenius)和原牛中东亚种(B. p. namadicus)独立驯化而来。原牛的所有亚种于距今9,000年至距今1,627年间陆续灭绝(Felius et al, 2014; Teletchea, 2019)。骨骼形态学、遗址的性别结构、有机物质同位素分析以及遗址中遗留的艺术形象表明: 普通牛驯化于距今10,300‒10,800年的新月沃土; 瘤牛最早驯化于距今8,000年的古代印度河流域文明遗址, 今为巴基斯坦俾路支省梅赫尔格尔(Chen et al, 2010; Felius et al, 2014)。家牛的独立驯化次数存在争议。非洲埃及、利比亚等地的考古遗址中都出现过牛遗骨, 且非洲家牛与欧亚家牛骨骼形态学差异很大, 线粒体单倍型谱系与核基因组分化程度高, 因此有学者认为原牛在非洲北部发生过独立驯化(陈宁博, 2019; Pitt et al, 2019)。研究表明, 非洲家牛高度分化的模式很可能是由于当地野生原牛对传播而来的驯化普通牛高比例的基因渗入导致(Loftus et al, 1994; Pitt et al, 2019; 李联萍等, 2020)。同样, 普通牛和瘤牛在扩散到亚洲的过程中也融入了牦牛、爪哇牛、大额牛等近缘种群, 这些丰富的遗传资源大大提升了家牛基因多样性与环境适应性(李联萍等, 2020)。古代家牛主要被用来祭祀和食用, 后用于农作劳役和产奶, 是农耕社会的主要畜力来源。在“以农为本”的古代中国, 牛肉在整个肉食资源中的比例始终不高, 《礼记》中曾记载“诸侯无故不杀牛”。
尽管家马对于我们现今的日常生活不那么重要, 但它在过去不仅提升了我们的贸易、通信、出行、文明传播的速度, 还从根本上改变了冷兵器时代的作战模式, 是国家宝贵的军事资源(Orlando, 2020a)。除人类外, 马的古代基因组是被发掘和研究的最为广泛的, 然而家马的驯化起源仍然未知(Orlando, 2020b)。线粒体基因组分析表明, 野马在家马驯化过程中贡献了73%的遗传多样性, 而野生祖先与近缘种群的濒临消失使得通过基因组亲缘关系来推断家马驯化起源的方法变得非常困难(陶克涛, 2021)。考古学证据曾将家马的驯化起源指向伊比利亚半岛和位于欧亚草原的哈萨克斯坦博泰文化遗址, 但分子遗传学表明, 前者较高的多态性更可能来源于野马频繁的基因渗入, 后者遗骸则是普氏野马的祖先(Orlando, 2020a; 陶克涛, 2021)。总之, 家马的驯化起源还需要学者们不断探索研究。
4 结语与展望
研究家养动物驯化起源, 不仅有助于了解复杂性状的遗传机制, 为解析动物重要性状相关基因提供了有效途径, 也对于了解人类自身的进化和社会发展具有重要的意义。同时, 研究家养动物驯化起源对保护野生动物资源和家养动物的种质资源创制提供理论指导和遗传材料。
考古学和分子遗传学从不同的角度为家养动物的驯化起源研究提供了证据。然而, 考古遗址及出土遗骸的稀缺导致能获得的直接证据有限; 分子和群体遗传学方法可以通过现代样本重建复杂的驯化历史, 但是其准确性和分辨率仍需要进一步验证。古DNA是连接考古学和分子遗传学的桥梁, 拉近了两方面的证据。未来动物驯化起源研究需要将人文地理、考古形态、古DNA, 以及分子和群体遗传多方面的证据融合, 并结合统计学和计算机技术以获得更全面和有效的信息。
致谢
感谢中国社会科学院袁靖先生对本文的指导!
参考文献
Adaptation and possible ancient inter species introgression in pigs identified by whole-genome sequencing
DOI:10.1038/ng.3199 URL [本文引用: 2]
Convergent genomic signatures of domestication in sheep and goats
DOI:10.1038/s41467-018-03206-y URL [本文引用: 1]
Fast model-based estimation of ancestry in unrelated individuals
DOI:10.1101/gr.094052.109
PMID:19648217
[本文引用: 1]

Population stratification has long been recognized as a confounding factor in genetic association studies. Estimated ancestries, derived from multi-locus genotype data, can be used to perform a statistical correction for population stratification. One popular technique for estimation of ancestry is the model-based approach embodied by the widely applied program structure. Another approach, implemented in the program EIGENSTRAT, relies on Principal Component Analysis rather than model-based estimation and does not directly deliver admixture fractions. EIGENSTRAT has gained in popularity in part owing to its remarkable speed in comparison to structure. We present a new algorithm and a program, ADMIXTURE, for model-based estimation of ancestry in unrelated individuals. ADMIXTURE adopts the likelihood model embedded in structure. However, ADMIXTURE runs considerably faster, solving problems in minutes that take structure hours. In many of our experiments, we have found that ADMIXTURE is almost as fast as EIGENSTRAT. The runtime improvements of ADMIXTURE rely on a fast block relaxation scheme using sequential quadratic programming for block updates, coupled with a novel quasi-Newton acceleration of convergence. Our algorithm also runs faster and with greater accuracy than the implementation of an Expectation-Maximization (EM) algorithm incorporated in the program FRAPPE. Our simulations show that ADMIXTURE's maximum likelihood estimates of the underlying admixture coefficients and ancestral allele frequencies are as accurate as structure's Bayesian estimates. On real-world data sets, ADMIXTURE's estimates are directly comparable to those from structure and EIGENSTRAT. Taken together, our results show that ADMIXTURE's computational speed opens up the possibility of using a much larger set of markers in model-based ancestry estimation and that its estimates are suitable for use in correcting for population stratification in association studies.
Deleterious alleles in the context of domestication, inbreeding, and selection
DOI:10.1111/eva.12691 URL [本文引用: 1]
Complex population structure in African village dogs and its implications for inferring dog domestication history
Phylogenetic distinctiveness of Middle Eastern and Southeast Asian village dog Y chromosomes illuminates dog origins
DOI:10.1371/journal.pone.0028496 URL [本文引用: 1]
The complex origins of domesticated crops in the Fertile Crescent
DOI:10.1016/j.tree.2008.09.008 URL [本文引用: 1]
Identity by descent between distant relatives: Detection and applications
DOI:10.1146/annurev-genet-110711-155534
PMID:22994355
[本文引用: 1]
Short segments of identity by descent (IBD) between individuals with no known relationship can be detected using genome-wide single nucleotide polymorphism data and recently developed statistical methodology. Emerging applications for the detected IBD segments include IBD mapping, haplotype phase inference, genotype imputation, and inference of population structure. In this review, we explain the principles behind methods for IBD segment detection, describe recently developed methods, discuss approaches to comparing methods, and give an overview of applications.
Research on the origins and spread of Chinese goat
中国山羊的起源与扩散研究
Whole-Genome Resequencing Reveals World-Wide Ancestry and Adaptive Introgression Events of Domesticated Cattle in East Asia
PhD dissertation,
全基因组重测序分析揭示东亚家牛的祖先与多重适应性基因渗入
博士学位论文,
Zebu cattle are an exclusive legacy of the south Asia Neolithic
DOI:10.1093/molbev/msp213 URL [本文引用: 1]
Genetic methods and applications in the study of the origin of domestic animals
家养动物起源研究的遗传学方法及其应用
Revealing the history of sheep domestication using retrovirus integrations
DOI:10.1126/science.1170587 URL [本文引用: 1]
Early Neolithic pig domestication at Jiahu, Henan Province, China: Clues from molar shape analyses using geometric morphometric approaches
DOI:10.1016/j.jas.2010.07.024 URL [本文引用: 4]
The hidden side of invasions: Massive introgression by local genes
Ancient goat genomes reveal mosaic domestication in the Fertile Crescent
DOI:10.1126/science.aas9411 URL [本文引用: 1]
Ancient genomics
Guns, Germs, and Steel: The Fates of Human Societies
Evolution, consequences and future of plant and animal domestication
DOI:10.1038/nature01019 URL [本文引用: 2]
Farmers and their languages: The first expansions
The largest movements and replacements of human populations since the end of the Ice Ages resulted from the geographically uneven rise of food production around the world. The first farming societies thereby gained great advantages over hunter-gatherer societies. But most of those resulting shifts of populations and languages are complex, controversial, or both. We discuss the main complications and specific examples involving 15 language families. Further progress will depend on interdisciplinary research that combines archaeology, crop and livestock studies, physical anthropology, genetics, and linguistics.
From wild animals to domestic pets, an evolutionary view of domestication
Identification of the yellow skin gene reveals a hybrid origin of the domestic chicken
DOI:10.1371/journal.pgen.1000010 URL [本文引用: 1]
Born free? New evidence for the status of Sus scrofa at Neolithic Cayonu Tepesi
DOI:10.3406/paleo.2001.4731 URL [本文引用: 2]
On the history of cattle genetic resources
DOI:10.3390/d6040705 URL [本文引用: 2]
The evolution of Suidae
DOI:10.1146/annurev-animal-021815-111155 URL [本文引用: 4]
Animal domestication in the era of ancient genomics
DOI:10.1038/s41576-020-0225-0 URL [本文引用: 5]
Ancient pigs reveal a near-complete genomic turnover following their introduction to Europe
Evidence of long-term gene flow and selection during domestication from analyses of Eurasian wild and domestic pig genomes
Frantz, Laurent A. F.; Madsen, Ole; Megens, Hendrik-Jan; Bosse, Mirte; Paudel, Yogesh; Crooijmans, Richard P. M. A.; Groenen, Martien A. M. Wageningen Univ, Anim Breeding & Genom Grp, NL-6700 AP Wageningen, Netherlands. Frantz, Laurent A. F.; Larson, Greger Univ Oxford, Res Lab Archaeol & Hist Art, Palaeogen & Bioarchaeol Res Network, Oxford, England. Schraiber, Joshua G. Univ Calif Berkeley, Dept Integrat Biol, Berkeley, CA 94720 USA. Schraiber, Joshua G. Univ Washington, Dept Genome Sci, Seattle, WA 98195 USA. Cagan, Alex Max Planck Inst Evolutionary Anthropol, Dept Evolutionary Genet, Leipzig, Germany.
DNA analysis of an early modern human from Tianyuan Cave, China
One subspecies of the red junglefowl (Gallus gallus gallus) suffices as the matriarchic ancestor of all domestic breeds
Monophyletic origin and unique dispersal patterns of domestic fowls
Single-stranded DNA library preparation for the sequencing of ancient or damaged DNA
DOI:10.1038/nprot.2013.038 URL [本文引用: 1]
Coming of age: Ten years of next-generation sequencing technologies
DOI:10.1038/nrg.2016.49
PMID:27184599
[本文引用: 1]
Since the completion of the human genome project in 2003, extraordinary progress has been made in genome sequencing technologies, which has led to a decreased cost per megabase and an increase in the number and diversity of sequenced genomes. An astonishing complexity of genome architecture has been revealed, bringing these sequencing technologies to even greater advancements. Some approaches maximize the number of bases sequenced in the least amount of time, generating a wealth of data that can be used to understand increasingly complex phenotypes. Alternatively, other approaches now aim to sequence longer contiguous pieces of DNA, which are essential for resolving structurally complex regions. These and other strategies are providing researchers and clinicians a variety of tools to probe genomes in greater depth, leading to an enhanced understanding of how genome sequence variants underlie phenotype and disease.
Analyses of pig genomes provide insight into porcine demography and evolution
DOI:10.1038/nature11622 URL [本文引用: 1]
Genetic diversity in farm animals-A review
DOI:10.1111/j.1365-2052.2010.02038.x URL [本文引用: 1]
Linear enamel hypoplasia as an indicator of physiological stress in great apes: Reviewing the evidence in light of enamel growth variation
DOI:10.1002/ajpa.21619
PMID:22610895
[本文引用: 1]
Physiological stress, such as malnutrition or illness, can disrupt normal enamel growth, resulting in linear enamel hypoplasias (LEHs). Although ecological factors may contribute to LEH expression, other factors, such as surface abrasion and enamel growth variables, are also likely to be involved. Attention to these other factors is necessary before we can begin to understand what LEH might signify in terms of ecological sources of physiological stress in non-human primates. This study focuses on assessing the contribution of these other factors to variation in LEH expression within and across great ape taxa. Here, we present LEH data from unabraded crown regions in samples of seven great ape species. We analyze these data with respect to lateral enamel formation time and the angles that striae of Retzius make with the enamel surface, as these variables are expected to affect variation in LEH expression. We find that although the duration of enamel formation is associated with sex differences in LEH expression, it is not clearly related to taxonomic variation in LEH expression, and does not explain the low frequency of LEH in mountain gorillas found in this and a previous study. Our data on striae of Retzius angles suggest that these influence LEH expression along the tooth crown and may contribute to the consistently high frequencies of LEH seen in Pongo in this and previous studies. We suggest that future work aimed at understanding species variation in these angles is crucial to evaluating taxonomic patterns of LEH expression in great apes.Copyright © 2012 Wiley Periodicals, Inc.
Prevalence and the duration of linear enamel hypoplasia: A comparative study of Neandertals and Inuit foragers
As a dental indicator of generalized physiological stress, enamel hypoplasia has been the subject of several Neandertal studies. While previous studies generally have found high frequencies of enamel hypoplasia in Neandertals, the significance of this finding varies with frequencies of enamel hypoplasia in comparative samples. The present investigation was undertaken to ascertain if the enamel hypoplasia evidence in Neandertals suggests a high level of physiological stress relative to a modern human foraging group, represented here by an archaeological sample of Inuit from Point Hope, Alaska. Unlike previous studies, this study focused specifically on linear enamel hypoplasia (LEH), emphasizing systemic over localized causes of this defect by considering LEH to be present in an individual only if LEH defects occur on two anterior teeth with overlapping crown formation periods. Moreover, this study is the first to evaluate the average growth disruption duration represented by these defects in Neandertals and a comparative foraging group. In the prevalence analysis, 7/18 Neandertal individuals (from Krapina and southern France) and 21/56 Neandertal anterior teeth were affected by LEH, or 38.9% and 37.5% respectively. These values do not differ significantly from those of the Inuit sample in which 8/21, or 38.1% of individuals, and 32/111, or 28.8% of anterior teeth were affected. For the growth disruption duration analysis, 22 defects representing separate episodes of growth disruption in Neandertals were compared with 22 defects in the Inuit group using three indicators of duration: the number of perikymata (growth increments) in the occlusal walls of LEH defects, the total number of perikymata within them, and defect width. Only one indicator, the total number of perikymata within defects, differed significantly between the Inuit and Neandertal groups (an average of 13.4 vs. 7.3 perikymata), suggesting that if there is any difference between them, the Inuit defects may actually represent longer growth disruptions than the Neandertal defects. Thus, while stress indicators other than linear enamel hypoplasia may eventually show that Neandertal populations were more stressed than those of modern foragers, the evidence from linear enamel hypoplasia does not lend support to this idea.
Domestic dog (Canis familiaris) diets among coastal Late Archaic groups of northeastern North America: A case study for the canine surrogacy approach
DOI:10.1016/j.jaa.2013.04.005 URL [本文引用: 2]
A genomic inference of the white Plymouth rock genealogy
DOI:10.3382/ps/pez411
PMID:31309227
[本文引用: 1]
Crossing of populations has been, and still is, a central component in domestication and breed and variety formation. It is a way for breeders to utilize heterosis and to introduce new genetic variation into existing plant and livestock populations. During the mid-19th century, several chicken breeds that had been introduced to America from Europe and Asia became the founders for those formed in the USA. Historical records about the genealogy of these populations are often unclear and inconsistent. Here, we used genomics in an attempt to describe the ancestry of the White Plymouth Rock (WPR) chicken. In total, 150 chickens from the WPR and 8 other stocks that historical records suggested contributed to its formation were whole-genome re-sequenced. The admixture analyses of the autosomal and sex chromosomes showed that the WPR was likely founded as a cross between a paternal lineage that was primarily Dominique, and a maternal lineage where Black Java and Cochin contributed in essentially equal proportions. These results were consistent and provided quantification with the historical records that they were the main contributors to the WPR. The genomic analyses also revealed genome-wide contributions (<10% each) by Brahma, Langshan, and Black Minorca. When viewed on an individual chromosomal basis, contributions varied considerably among stocks.© 2019 Poultry Science Association Inc.
Massive migration from the steppe was a source for Indo-European languages in Europe
DOI:10.1038/nature14317 URL [本文引用: 1]
Amplification of mitochondrial DNA fragments from ancient human teeth and bones
Domestication revisited: Its implications for faunal analysis
Molecular-clock methods for estimating evolutionary rates and timescales
DOI:10.1111/mec.12953 URL [本文引用: 1]
Pig domestication in ancient China
DOI:10.1017/S0003598X00091171 URL [本文引用: 1]
The Neolithic transition in Vietnam: Assessing evidence for early pig management and domesticated dog
DOI:10.1016/j.jasrep.2019.102042 URL [本文引用: 1]
mtDNA analysis reveals enriched pathogenic mutations in Tibetan highlanders
DOI:10.1038/srep31083 URL [本文引用: 1]
How culture shaped the human genome: Bringing genetics and the human sciences together
DOI:10.1038/nrg2734
PMID:20084086
[本文引用: 1]
Researchers from diverse backgrounds are converging on the view that human evolution has been shaped by gene-culture interactions. Theoretical biologists have used population genetic models to demonstrate that cultural processes can have a profound effect on human evolution, and anthropologists are investigating cultural practices that modify current selection. These findings are supported by recent analyses of human genetic variation, which reveal that hundreds of genes have been subject to recent positive selection, often in response to human activities. Here, we collate these data, highlighting the considerable potential for cross-disciplinary exchange to provide novel insights into how culture has shaped the human genome.
A population genetics view of animal domestication
DOI:10.1016/j.tig.2013.01.003 URL [本文引用: 6]
Phylogeny and ancient DNA of Sus provides insights into Neolithic expansion in Island Southeast Asia and Oceania
Worldwide phylogeography of wild boar reveals multiple centers of pig domestication
DOI:10.1126/science.1106927 URL [本文引用: 2]
The evolution of animal domestication
DOI:10.1146/annurev-ecolsys-110512-135813 URL [本文引用: 6]
Patterns of East Asian pig domestication, migration, and turnover revealed by modern and ancient DNA
Current perspectives and the future of domestication studies
In search of the wild chicken
DOI:10.1126/science.338.6110.1020 URL [本文引用: 1]
Worldwide human relationships inferred from genome-wide patterns of variation
DOI:10.1126/science.1153717 URL [本文引用: 1]
Advance in research on whole-genome genetic diversity and origins in cattle
家牛全基因组遗传多样性与起源研究进展
Genomic analysis on pygmy hog reveals extensive interbreeding during wild boar expansion
DOI:10.1038/s41467-019-10017-2 URL [本文引用: 1]
Multiple maternal origins of chickens: Out of the Asian jungles
DOI:10.1016/j.ympev.2005.09.014 URL [本文引用: 1]
Evidence for two independent domestications of cattle
Domestication is not an ancient moment of selection for prosociality: Insights from dogs and modern humans
Early development of domestic pig breeding in China
中国家猪饲养的早期发展
Research on the origin of Chinese domestic pig
中国家猪起源机制蠡测
Restudy of the pigs’ bones from the Jiahu site in Wuyang County, Henan
河南舞阳县贾湖遗址出土猪骨的再研究
Mitogenomic meta-analysis identifies two phases of migration in the history of eastern Eurasian sheep
DOI:10.1093/molbev/msv139 URL [本文引用: 1]
Taming the past: Ancient DNA and the study of animal domestication
DOI:10.1146/annurev-animal-022516-022747
PMID:27813680
[本文引用: 2]
During the last decade, ancient DNA research has been revolutionized by the availability of increasingly powerful DNA sequencing and ancillary genomics technologies, giving rise to the new field of paleogenomics. In this review, we show how our understanding of the genetic basis of animal domestication and the origins and dispersal of livestock and companion animals during the Upper Paleolithic and Neolithic periods is being rapidly transformed through new scientific knowledge generated with paleogenomic methods. These techniques have been particularly informative in revealing high-resolution patterns of artificial and natural selection and evidence for significant admixture between early domestic animal populations and their wild congeners.
Multiplexed DNA sequence capture of mitochondrial genomes using PCR products
DOI:10.1371/journal.pone.0014004 URL [本文引用: 1]
Chicken domestication: An updated perspective based on mitochondrial genomes
DOI:10.1038/hdy.2012.83
PMID:23211792
[本文引用: 1]
Domestic chickens (Gallus gallus domesticus) fulfill various roles ranging from food and entertainment to religion and ornamentation. To survey its genetic diversity and trace the history of domestication, we investigated a total of 4938 mitochondrial DNA (mtDNA) fragments including 2843 previously published and 2095 de novo units from 2044 domestic chickens and 51 red junglefowl (Gallus gallus). To obtain the highest possible level of molecular resolution, 50 representative samples were further selected for total mtDNA genome sequencing. A fine-gained mtDNA phylogeny was investigated by defining haplogroups A-I and W-Z. Common haplogroups A-G were shared by domestic chickens and red junglefowl. Rare haplogroups H-I and W-Z were specific to domestic chickens and red junglefowl, respectively. We re-evaluated the global mtDNA profiles of chickens. The geographic distribution for each of major haplogroups was examined. Our results revealed new complexities of history in chicken domestication because in the phylogeny lineages from the red junglefowl were mingled with those of the domestic chickens. Several local domestication events in South Asia, Southwest China and Southeast Asia were identified. The assessment of chicken mtDNA data also facilitated our understanding about the Austronesian settlement in the Pacific.
Statistical guidelines for detecting past population shifts using ancient DNA
DOI:10.1093/molbev/mss094
PMID:22427706
[本文引用: 1]
Populations carry a genetic signal of their demographic past, providing an opportunity for investigating the processes that shaped their evolution. Our ability to infer population histories can be enhanced by including ancient DNA data. Using serial-coalescent simulations and a range of both quantitative and temporal sampling schemes, we test the power of ancient mitochondrial sequences and nuclear single-nucleotide polymorphisms (SNPs) to detect past population bottlenecks. Within our simulated framework, mitochondrial sequences have only limited power to detect subtle bottlenecks and/or fast post-bottleneck recoveries. In contrast, nuclear SNPs can detect bottlenecks followed by rapid recovery, although bottlenecks involving reduction of less than half the population are generally detected with low power unless extensive genetic information from ancient individuals is available. Our results provide useful guidelines for scaling sampling schemes and for optimizing our ability to infer past population dynamics. In addition, our results suggest that many ancient DNA studies may face power issues in detecting moderate demographic collapses and/or highly dynamic demographic shifts when based solely on mitochondrial information.
Mathematical model for studying genetic variation in terms of restriction endonucleases
Insights on patterns of developmental disturbances from the analysis of linear enamel hypoplasia in a Neolithic sample from Liguria (northwestern Italy)
DOI:S1879-9817(19)30152-4
PMID:31901428
[本文引用: 2]
To assess developmental disturbances through the analysis of linear enamel hypoplasia (LEH) frequency and to infer environmental stress and life history within Neolithic communities from Liguria (Italy).43 unworn/minimally worn permanent anterior teeth of 13 individuals recovered from nearby caves and dated to c. 4800-4400 cal. BCE.LEH defects were identified with high-resolution macrophotos of dental replicas, age at LEH was calculated via perikymata counts. LEH defects matched between two or more teeth were considered as systemic disturbances. LEH frequency by age classes was analyzed via GLZ and Friedman ANOVA.Number of matched defects per individual range between 2-12. The mean LEH per individual was highest in the 2.5-2.99 age category, with a significant increase relative to earlier growth stages, followed by a decline.LEH may reflect life-history in the local ecology of Neolithic Liguria, where several individuals with osteoarticular tuberculosis have been recorded. Disease burden may have triggered developmental disturbances around the time of weaning. Age at first defect was negatively correlated with age at death and positively with the total number of defects, suggesting that early stress may have affected survivorship.The study contributes to the reconstruction of ecological pressures among Neolithic people of Liguria, and informs on environmental challenges during the Neolithic adaptive expansion.The visual examination of macrophotos is prone to observer error; mid-crown tends to display more visible LEH due to tooth architecture.Apply different quantitative methods to examine severity and duration of disturbances.Copyright © 2020 Elsevier Inc. All rights reserved.
Ancient genomes reveal unexpected horse domestication and management dynamics
The evolutionary and historical foundation of the modern horse: Lessons from ancient genomics
DOI:10.1146/annurev-genet-021920-011805 URL [本文引用: 1]
Using ancient DNA to understand evolutionary and ecological processes
DOI:10.1146/annurev-ecolsys-120213-091712 URL [本文引用: 1]
Recalibrating Equus evolution using the genome sequence of an early Middle Pleistocene horse
DOI:10.1038/nature12323 URL [本文引用: 1]
Inferring the age of a fixed beneficial allele
Ancient DNA and the polymerase chain reaction. The emerging field of molecular archaeology
Genome sequencing of the extinct Eurasian wild aurochs, Bos primigenius, illuminates the phylogeography and evolution of cattle
DOI:10.1186/s13059-015-0790-2 URL [本文引用: 1]
Ancient admixture in human history
DOI:10.1534/genetics.112.145037
PMID:22960212
[本文引用: 2]
Population mixture is an important process in biology. We present a suite of methods for learning about population mixtures, implemented in a software package called ADMIXTOOLS, that support formal tests for whether mixture occurred and make it possible to infer proportions and dates of mixture. We also describe the development of a new single nucleotide polymorphism (SNP) array consisting of 629,433 sites with clearly documented ascertainment that was specifically designed for population genetic analyses and that we genotyped in 934 individuals from 53 diverse populations. To illustrate the methods, we give a number of examples that provide new insights about the history of human admixture. The most striking finding is a clear signal of admixture into northern Europe, with one ancestral population related to present-day Basques and Sardinians and the other related to present-day populations of northeast Asia and the Americas. This likely reflects a history of admixture between Neolithic migrants and the indigenous Mesolithic population of Europe, consistent with recent analyses of ancient bones from Sweden and the sequencing of the genome of the Tyrolean "Iceman."
Questioning new answers regarding Holocene chicken domestication in China
Optimal ancient DNA yields from the inner ear part of the human petrous bone
DOI:10.1371/journal.pone.0129102 URL [本文引用: 1]
Domestication of cattle: Two or three events?
DOI:10.1111/eva.12674 URL [本文引用: 2]
Archaeological dogs from the Early Holocene Zhokhov site in the Eastern Siberian Arctic
DOI:10.1016/j.jasrep.2017.04.003 URL [本文引用: 1]
Pigs: A Handbook to the Breeds of the World. Comstock Publishing Associates
Inference of population structure using multilocus genotype data
DOI:10.1093/genetics/155.2.945
PMID:10835412
[本文引用: 1]
We describe a model-based clustering method for using multilocus genotype data to infer population structure and assign individuals to populations. We assume a model in which there are K populations (where K may be unknown), each of which is characterized by a set of allele frequencies at each locus. Individuals in the sample are assigned (probabilistically) to populations, or jointly to two or more populations if their genotypes indicate that they are admixed. Our model does not assume a particular mutation process, and it can be applied to most of the commonly used genetic markers, provided that they are not closely linked. Applications of our method include demonstrating the presence of population structure, assigning individuals to populations, studying hybrid zones, and identifying migrants and admixed individuals. We show that the method can produce highly accurate assignments using modest numbers of loci-e.g., seven microsatellite loci in an example using genotype data from an endangered bird species. The software used for this article is available from http://www.stats.ox.ac.uk/ approximately pritch/home. html.
Reconstructing Indian population history
DOI:10.1038/nature08365 URL [本文引用: 1]
Distinguishing wild boar from domestic pigs in prehistory: A review of approaches and recent results
DOI:10.1007/s10963-012-9055-0 URL [本文引用: 1]
Approaching sheep herds origins and the emergence of the wool economy in continental Europe during the Bronze Age
DOI:10.1007/s12520-019-00856-x
[本文引用: 1]
In recent years, extensive archaeological studies have provided us with new knowledge on wool and woollen textile production in continental Europe during the Bronze Age. Concentrations of large numbers of textile tools, and of zooarchaeological evidence suggesting intense sheepherding, hint at specialized centres of wool production during the Bronze Age. The aim of this paper is to discuss whether engagement with this economic activity was facilitated by the introduction of new foreign sheep types, possibly from the Eastern Mediterranean, where well-established wool economies existed, or by using local sheep, or a mixture of local and non-local types. A small-scale genetic pilot study, presented in this paper, primarily aimed at testing the DNA preservation, and thus the genomic potential in Bronze Age sheep remains provides evidence of both mitochondrial haplogroups A and B among Bronze Age sheep in Hungary. This result could hint at sheep herds with mixed origin but further in-depth studies are necessary to address this. We aim to promote scholarly interest in the issue and propose new directions for research on this topic.
Almost 20 years of Neanderthal palaeogenetics: Adaptation, admixture, diversity, demography and extinction
Powerful inference with the D-statistic on low-coverage whole-genome data
DOI:10.1534/g3.117.300192 URL [本文引用: 1]
Approximate Bayesian computation
DOI:10.1371/journal.pcbi.1002803 URL [本文引用: 1]
Statistical method for testing the neutral mutation hypothesis by DNA polymorphism
DOI:10.1093/genetics/123.3.585
PMID:2513255
[本文引用: 1]
The relationship between the two estimates of genetic variation at the DNA level, namely the number of segregating sites and the average number of nucleotide differences estimated from pairwise comparison, is investigated. It is found that the correlation between these two estimates is large when the sample size is small, and decreases slowly as the sample size increases. Using the relationship obtained, a statistical method for testing the neutral mutation hypothesis is developed. This method needs only the data of DNA polymorphism, namely the genetic variation within population at the DNA level. A simple method of computer simulation, that was used in order to obtain the distribution of a new statistic developed, is also presented. Applying this statistical method to the five regions of DNA sequences in Drosophila melanogaster, it is found that large insertion/deletion (greater than 100 bp) is deleterious. It is suggested that the natural selection against large insertion/deletion is so weak that a large amount of variation is maintained in a population.
Study on Genetic Diversity of Asian Domestic Horse--Examples from Chinese and Mongolian Populations
PhD dissertation,
亚洲家马遗传多样性的研究--以中国和蒙古国种群为例
博士学位论文,
Domesticated landscapes: The subsistence ecology of plant and animal domestication
DOI:10.1023/B:JARM.0000005510.54214.57 URL [本文引用: 1]
Whole genome sequence analysis reveals genetic structure and X-chromosome haplotype structure in indigenous Chinese pigs
DOI:10.1038/s41598-020-66061-2 URL [本文引用: 1]
Heterogeneous distribution of SNPs in the human genome: Microsatellites as predictors of nucleotide diversity and divergence
DOI:10.1016/j.ygeno.2009.12.003 URL [本文引用: 1]
Genome-wide analysis reveals artificial selection on coat colour and reproductive traits in Chinese domestic pigs
DOI:10.1111/1755-0998.12311 URL [本文引用: 1]
Domestication genomics: Evidence from animals
DOI:10.1146/annurev-animal-022513-114129 URL [本文引用: 2]
Out of southern East Asia: The natural history of domestic dogs across the world
DOI:10.1038/cr.2015.147 URL [本文引用: 1]
Local ancestry inference in large pedigrees
DOI:10.1038/s41598-019-57039-w URL [本文引用: 1]
863 genomes reveal the origin and domestication of chicken
DOI:10.1038/s41422-020-0349-y URL [本文引用: 2]
Large-scale genomic analysis reveals the genetic cost of chicken domestication
DOI:10.1186/s12915-021-01052-x URL [本文引用: 1]
Progress on animal domestication under population genetics
群体遗传学下动物驯化研究进展
Did chickens go North? New evidence for domestication
DOI:10.1016/0305-4403(88)90080-5 URL [本文引用: 1]
From globalized pig breeds to capitalist pigs: A study in animal cultures and evolutionary history
DOI:10.1093/envhis/emq143 URL [本文引用: 1]
Diverse plant and animal genetic records from Holocene and Pleistocene sediments
Genetic analyses of permafrost and temperate sediments reveal that plant and animal DNA may be preserved for long periods, even in the absence of obvious macrofossils. In Siberia, five permafrost cores ranging from 400,000 to 10,000 years old contained at least 19 different plant taxa, including the oldest authenticated ancient DNA sequences known, and megafaunal sequences including mammoth, bison, and horse. The genetic data record a number of dramatic changes in the taxonomic diversity and composition of Beringian vegetation and fauna. Temperate cave sediments in New Zealand also yielded DNA sequences of extinct biota, including two species of ratite moa, and 29 plant taxa characteristic of the prehuman environment. Therefore, many sedimentary deposits may contain unique, and widespread, genetic records of paleoenvironments.
Population phylogenomic analysis of mitochondrial DNA in wild boars and domestic pigs revealed multiple domestication events in East Asia
DOI:10.1186/gb-2007-8-11-r245 URL [本文引用: 1]
Systematic review on local ancestor inference from a mathematical and algorithmic perspective
DOI:10.3389/fgene.2021.639877 URL [本文引用: 1]
Early Holocene chicken domestication in Northern China
The reexamination on the origination of the domestication of animals in China
中国家养动物起源的再思考
Restudy of the origin of domestic chicken in ancient China
中国古代家鸡起源的再研究
Archaeogenetic analysis of Neolithic sheep from Anatolia suggests a complex demographic history since domestication
DOI:10.1038/s42003-021-02794-8
PMID:34773064
[本文引用: 2]
Sheep were among the first domesticated animals, but their demographic history is little understood. Here we analyzed nuclear polymorphism and mitochondrial data (mtDNA) from ancient central and west Anatolian sheep dating from Epipaleolithic to late Neolithic, comparatively with modern-day breeds and central Asian Neolithic/Bronze Age sheep (OBI). Analyzing ancient nuclear data, we found that Anatolian Neolithic sheep (ANS) are genetically closest to present-day European breeds relative to Asian breeds, a conclusion supported by mtDNA haplogroup frequencies. In contrast, OBI showed higher genetic affinity to present-day Asian breeds. These results suggest that the east-west genetic structure observed in present-day breeds had already emerged by 6000 BCE, hinting at multiple sheep domestication episodes or early wild introgression in southwest Asia. Furthermore, we found that ANS are genetically distinct from all modern breeds. Our results suggest that European and Anatolian domestic sheep gene pools have been strongly remolded since the Neolithic.© 2021. The Author(s).
Domestication and early agriculture in the Mediterranean Basin:Origins, diffusion, and impact
The domestication of animals
DOI:10.3998/jar.0521004.0068.201 URL [本文引用: 1]
Core questions in domestication research
A conversation on agricultural origins
DOI:10.1086/605553 URL [本文引用: 1]
A history of domesticated animals
DOI:10.2307/2363 URL [本文引用: 1]
Genomic variation in Pekin duck populations developed in three different countries as revealed by whole-genome data
DOI:10.1111/age.12639
PMID:29388248
[本文引用: 1]
It is well known that both British and American Pekin ducks originated from China. However, the populations differ substantially in production performance, but the genetic changes involved are still poorly understood. Herein, we sequenced 24 individual Pekin ducks (eight from each population) with an average sequencing depth of more than 45× for each population (mean coverage of 6.29 per individual). Among these populations from three different countries, we identified a large number of SNPs and indels as well as many unique population variants, which can be used as population-specific molecular markers. Genomic comparisons among the three duck populations revealed many candidate genes as well as pathways and Gene Ontology categories that are putatively associated with meat yield in the British population, growth in the American population and brain development in all three populations. These findings will enable a better understanding of the artificial selection history of Pekin ducks and provide a valuable resource for future research on the breeding of this species.© 2018 Stichting International Foundation for Animal Genetics.
Genomic reconstruction of the history of native sheep reveals the peopling patterns of nomads and the expansion of early pastoralism in East Asia
DOI:10.1093/molbev/msx181 URL [本文引用: 1]
Population structure and genetic introgression from wild relatives in worldwide goat populations
PhD dissertation.
世界山羊群体遗传结构及其野生近缘种基因渗入研究
博士学位论文,

{kind=link}
{kind=link}
{kind=link}
{kind=link}