


由于人类活动所引起的全球环境变化例如氮沉降、增温、增雨等问题已成为人们关注的焦点(Gilling et al, 2019)。大部分草地位于生态脆弱带上, 对气候和环境变化十分敏感(Li et al, 2005; 孙良杰等, 2012)。内蒙古草原占我国总草地面积的20%以上, 是欧亚草原的重要组成部分。该地区不仅是我国重要的牲畜和饲料生产基地, 而且有着不可替代的生态系统功能(如: 碳固定、维持和保护生物多样性等) (Ling et al, 2017)。因此, 研究全球变化对我国内蒙古草地生态系统的影响, 对于准确把握全球变化背景下陆地生态系统的响应和适应机制具有重要意义, 同时可为该地区制定相应的生态环境政策提供依据(贺纪正和郭良栋, 2013)。
土壤微生物是土壤有机质和养分转化与循环的主要参与者与调节者, 在土壤生态系统中起着重要作用(Bardgett & van der Putten, 2014; Thakur & Geisen, 2019)。研究土壤微生物多样性能够更敏感地探知全球变化背景下生态系统的变化及其响应机制, 并为阐明生态系统对气候变化的反馈机理提供依据。之前关注的重点多集中在全球环境变化因子对地上植物多样性及其功能的影响, 而对全球环境变化因子对地下生物尤其是微生物多样性影响的研究尚重视不够(Rilling et al, 2019)。随着分子生物学技术的发展和普及, 人们在环境变化对微生物多样性的影响方面进行了大量研究。本文针对目前关于我国内蒙古草地生态系统微生物多样性维持机制的相关研究进展进行综述, 以期为未来相关研究的继续深入提供一定的参考。
1 各种环境变化对土壤微生物群落的相对影响大小的比较
人类活动引起多种环境因子的变化, 例如: 氮沉降、生物多样性丧失、增温、增雨等(Balint et al, 2011; 孙良杰等, 2012; IPCC, 2014; Rillig et al, 2019)。目前, 环境变化对土壤微生物群落影响的研究多集中在单因素研究, 缺少同时考虑多种因素的系统性研究(Rillig et al, 2019)。不同环境变化类型对土壤微生物的影响不同。研究者已通过多角度分析并探讨了单因素对地下微生物群落的影响(孙良杰等, 2012; Zhou et al, 2017; Ren et al, 2018; Wang C et al, 2018; Jansson & Hofmockel, 2020), 这里就不再详述。已有的多因素研究表明, 不同环境变化因子的相对影响大小不同。在内蒙古半干旱草原, Ling等(2017)通过比较长期(17年)施氮和施磷对土壤细菌群落的影响, 发现氮添加速率与细菌Chao指数和Shannon指数都呈显著负相关, 磷添加仅与细菌群落的Chao指数显著负相关; 与磷添加相比, 地下细菌群落对氮添加更敏感。9年的施氮和增雨实验表明, 施氮显著降低, 增雨显著增加细菌丰富度、Shannon指数、Chao指数和Faith’s phylogenetic指数, 施氮作用是影响细菌群落β多样性的最主要影响因素(Li et al, 2016)。在荒漠草原, 施氮和水分调控显著降低了0-2 cm土壤可培养细菌群落丰富度(Jia et al, 2019)。Zhang等(2016a)在内蒙古典型草原同时考虑氮沉降、植物多样性丧失、放牧、磷素添加、增温、增雨等, 单因子及多因子之间的组合, 共计16种环境变化对土壤微生物多样性的影响(图1), 发现与别的环境变化相比, 氮沉降及其与其他因素的结合具有更大的影响作用。与草原生态系统类似, 在农田生态系统中, 与磷添加和钾添加相比, 氮添加处理对微生物群落有更大影响(Yu et al, 2019)。
图1
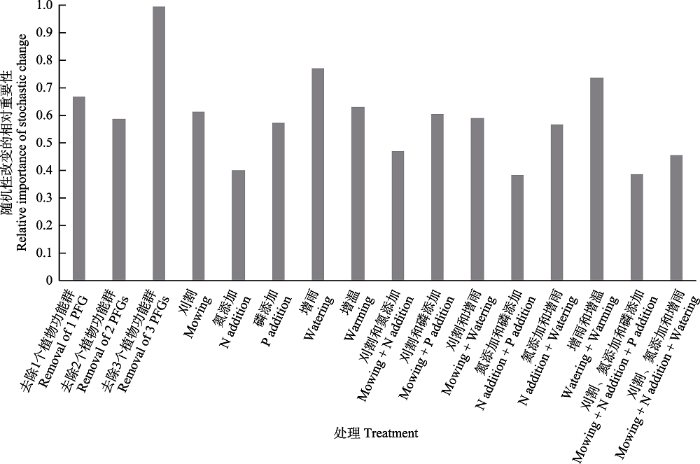
图1
随机性改变在16种环境变化下的相对重要性(改编自Zhang et al, 2016a)。如果随机改变的相对重要性 > 0.5, 说明环境改变主要调节随机性过程。PFG: 植物功能群。
Fig. 1
The relative importance of the stochastic change under 16 environmental changes (adapted from Zhang et al, 2016a). If the relative importance of the stochastic change is larger than 0.5, it means that stochastic processes are the primarily mediated processes. PFG, Plant functional groups.
研究者还比较了不同环境变化对功能菌群的影响。在内蒙古多伦典型草原, 同时考虑氮添加、磷添加及放牧对氨氧化古菌(AOA)、氨氧化细菌(AOB)群落丰度与结构的影响, 研究发现, 氮添加和放牧都对AOA和AOB有显著影响, 而磷添加处理只对AOB丰度有显著影响(Chen et al, 2014a)。9年的放牧、氮添加和增雨多因子研究表明, 氮添加对AOB和包含nosZ的反硝化群落结构有显著影响, 但增雨仅影响AOA的群落结构, 放牧对氮循环功能群基因的群落组成没有显著影响(Zhang CJ et al, 2018)。系统地比较各种氮循环微生物功能群(固氮、矿化、硝化和反硝化)对16种环境变化(氮沉降、植物多样性丧失、放牧、磷素添加、增温、增雨等环境变化因子及其组合)的响应, 发现所有功能群都对氮沉降敏感, 同时氨氧化细菌对各种环境变化敏感, 是各种环境变化的指示微生物(Zhang et al, 2013b)。该发现在其他生态系统中得到进一步验证(van Dorst et al, 2014; Eo & Park, 2016)。综合已经完成的研究, 在内蒙古草原各种环境变化类型中, 氮沉降的影响最大, 增雨、增温、植物多样性丧失也有一定影响, 而放牧和磷素添加的影响较小(Zhang et al, 2013a, c, 2014a, 2017a)。
丛枝菌根真菌(AMF)是土壤微生物的重要组成部分, 与超过80%的陆地植物构建共生体系(Smith & Read, 2008; Wang DL et al, 2018)。在内蒙古多伦典型草原, 施氮主要改变了AMF的群落组成, 施磷主要影响AMF的多度, 增温则显著增加AMF的OTU (operational taxonomic unit)丰富度(Chen et al, 2014b, 2017; Kim et al, 2015)。6年的增雨和增温研究表明, 与增温相比, 增雨对AMF群落的影响更大: 增雨显著降低AMF的α多样性(OTU丰富度及Shannon指数), 而增加其β多样性, 并显著影响其群落组成(Gao et al, 2016)。同时研究增雨和施氮对丛枝菌根真菌群落的影响发现, 与施氮相比, 由增雨引起的土壤水分可利用性的变化对AMF群落的影响更大(Li et al, 2015)。因此, 不同环境变化因子对AMF群落的影响方式不同, 增雨可能影响最大, 这需要从多因子角度进行深入研究。
2 各环境变化影响土壤微生物群落多样性的物理化学机制
不同环境变化对土壤微生物的影响不同, 且其影响的相对大小也不同, 这是由于各种环境变化影响的内在物理化学机制的不同所造成的。
模型预测, 未来氮沉降仍会持续增加(Dentener et al, 2006; Galloway et al, 2008)。施氮一方面通过提高土壤有效氮含量, 促进土壤微生物生长, 影响其群落组成; 另一方面, 施氮会降低土壤pH值, 造成土壤盐基阳离子(Mg2+, Ca2+, Na+)流失和Al3+含量增高, 进而影响土壤微生物群落(Zhou et al, 2017)。在全球尺度上, 氮沉降主要通过增加土壤氮可利用性而非降低土壤pH值影响土壤微生物群落组成(Zhou et al, 2017)。与全球结果不同, 在内蒙古草原, 氮沉降主要通过降低土壤pH值影响土壤微生物群落。在内蒙古典型草原, 氮添加导致的土壤pH值的降低是引起土壤细菌群落多样性、相对丰度及主要细菌门类群落多样性变化的首要因素(Zhang et al, 2011, 2013c, 2014b; Zhang & Han, 2012)。
降水主要通过增加土壤水分和提高土壤pH值影响土壤微生物群落。降水能缓解氮沉降对土壤微生物群落的影响, 有利于提高群落稳定性。Zhang等(2014b)研究氮沉降、增雨和放牧3个因素对土壤微生物的影响时发现, 降雨增加刺激了反硝化作用, 因而提高了土壤pH值; 因此降雨增加可以缓冲氮沉降对土壤pH值的降低, 部分抵消氮沉降对细菌群落的负作用(Zhang et al, 2014b)。9年的增雨和施氮梯度研究表明, 增雨不但削弱氮沉降对细菌群落α多样性的负作用, 而且缓解了氮沉降对细菌群落β多样性的影响, 因此可提高微生物群落稳定性。这可能是由于增雨增加淋溶作用并促进植物和微生物对土壤氮的吸收, 进而减少土壤中无机氮的积累, 从而缓解氮沉降对土壤微生物的影响(Li et al, 2016)。在呼伦贝尔贝加尔针茅草原, 高浓度梯度的氮沉降速率(> 100 kg N·ha-1·yr-1)显著增加真菌群落多样性, 但增雨减小了氮沉降对真菌群落的影响, 使真菌多样性不变, 因此增雨有助于氮沉降条件下真菌多样性的维持(Zhang HF et al, 2018)。
明确土壤微生物对气候变暖的反馈方向和机制是预测整个生态系统反应的关键。一方面增温条件下微生物酶的活力提高, 进而提高其对土壤有机质的分解能力, 向大气中释放更多的CO2和CH4, 从而对气候变暖造成正反馈(Heimann & Reichstein, 2008; Mackelprang et al, 2011; Karhu et al, 2014; McCalley et al, 2014)。另一方面, 微生物自身适应气候变暖, 从而对其无反馈或负反馈(Zhou et al, 2012)。然而, 由于土壤有机质成分复杂、大部分分解缓慢、微生物种类繁多等因素, 导致很难判别微生物对气候变暖的反馈方向和机制(Curtis et al, 2002; Handelsman et al, 2007)。Zhang等(2017b)对内蒙古典型草原上一个长期模拟增雨增温实验的土壤微生物群落进行了针对所有微生物基因组的高通量测序, 发现增温和增雨有利于微生物的生存, 促进它们对氮和硫的吸收与同化代谢, 增加群落的生物量和复杂性, 增强植物群落与微生物群落之间的紧密关系, 同时促进微生物对土壤难分解有机质的分解代谢。总而言之, 结合宏基因组证据与土壤物理化学指标, 发现气候变暖可提高典型草原土壤微生物群落分解土壤有机质的能力, 此为微生物对气候变暖正反馈的基因水平的最直接证据。除非降雨与气温同时增加, 否则在气候变暖条件下我国北方典型草原将会成为碳源。
由于人类活动所引起的环境变化如氮沉降、全球变暖等正在加速地球生物多样性的丧失(Dirzo & Raven, 2003; Balint et al, 2011; Chen et al, 2019)。植物作为微生物生长所需底物的供应者, 植物多样性丧失对微生物群落的影响至少有两种机制: 一方面, 植物多样性丧失导致群落生产力下降, 进而减少进入土壤的有机质的数量, 并最终影响微生物的生长及群落结构(Tilman, 1982; Tilman et al, 1996; Waldrop et al, 2006; Chen et al, 2019); 另一方面, 植物多样性的丧失降低了进入土壤有机质的种类及土壤微环境的多样性和组成, 进而影响微生物群落(Lodge, 1997; Hooper et al, 2000; Brodie et al, 2003; Chen et al, 2019)。一般认为植物多样性的丧失主要通过碳源多样性降低来减少土壤微生物多样性。但是该假设的支持证据并不多, 其生态学机理也不清楚。Zhang等(2017a)以内蒙古草原上一个长期的植物功能群去除实验为依托, 设置4个功能群多样性梯度(0-3), 用宏基因组技术测量微生物的分类学和功能基因多样性。结果发现实验处理没有影响微生物物种多样性, 但降低了功能基因多样性; 由植物多样性丧失造成的植物生产力降低是微生物功能基因多样性降低的主要因素, 而非植物多样性丧失本身。生产力降低导致供给微生物的新鲜易分解碳源减少, 进而增加了生态筛选过程的选择压力, 有利于负责能量生产等相关功能的基因, 同时不利于其他功能基因。总之, 该研究发现地上地下群落之间最关键的链接是碳源量影响微生物功能基因多样性, 而非植物多样性影响微生物分类多样性。
3 各环境变化影响的生态学机制
3.1 确定性过程与随机性过程的相对重要性
确定性过程(如种间竞争和环境过滤)和随机性过程(如物种扩散和生态漂变)在维持生物多样性中的相对重要性是生态学研究的一个核心问题。基于Bass-Becking在1934年提出的观点: “Everything is everywhere, but, the environment selects”, 一般认为, 微生物群落多样性及组成主要受到确定性过程的影响(de Wit & Bouvier, 2006; Hanson et al, 2012)。但越来越多证据表明, 随机性过程在微生物群落构建中也起到关键性作用(Caruso et al, 2011; Hao et al, 2016; Zhang et al, 2016a; Wang et al, 2017)。如今, 越来越多生态学家认为确定性过程和随机性过程共同决定微生物群落的构建(Cao & He, 2015; Wang et al, 2017; Shi et al, 2018)。
人类活动所引起的环境变化, 如氮沉降、全球变暖等可能通过调控确定性过程和随机性过程影响微生物群落的构建。换句话说, 在未经干扰的生态系统中, 确定性过程和随机性过程共同作用决定土壤微生物群落的构建, 环境变化可能会促进或者限制二者的作用。基于内蒙古典型草原上的一个长期模拟氮沉降实验, Zhang等(2011)利用直接计算的方法有效分离氮沉降引起的群落组成变异的确定性成分和随机性成分, 并且计算两者的相对重要性(百分比)。直接计算方法简单地说, 就是对于每种处理, 随机性变化 = (处理之间的平均组成变异) ‒ (对照之间的平均组成变异), 确定性变化 = (对照与处理之间的平均组成变异) ‒ (对照之间的平均组成变异)。该方法的前提条件是不同重复之间的群落组成变异主要是由随机性过程造成。该方法成功地应用于植物、细菌和氨氧化古菌群落构建的分析。研究发现, 随着氮添加水平的增加, 随机性过程的相对重要性呈现非线性下降。氮沉降通过调节随机过程的相对重要性来改变植物和微生物多样性, 因此需要根据不同的氮沉降速率和群落属性采取不同的多样性保护策略。Zhang等(2016a)应用同样的方法分析了典型草原上的16种不同的环境变化处理(植物多样性丧失、生物量收割、氮沉降、磷素添加、增雨、增温, 以及它们的共同作用), 结果发现在几乎所有的环境变化条件下, 随机过程变化的绝对值大于确定性过程变化的绝对值, 即这些环境变化主要是通过调节随机性变化而非确定性变化来影响土壤微生物群落(图1)。此外研究者用Chase (2010)的随机模拟方法证明了直接计算方法的正确性。需要注意的是, 环境变化主要调节了随机性变化, 包括促进和抑制两种情况。如果是抑制, 意味着随机性过程的重要性降低, 而确定性过程的重要性增加(Zhang et al, 2016a)。6年的增雨和施氮研究表明, 增雨增加了AMF群落构建中随机过程的重要性(Gao et al, 2016)。但也有研究表明, 增雨和施氮对AMF群落的构建机制没有显著影响, 环境过滤作用一直是其群落构建的主要机制(Chen et al, 2017)。此外, 随机性过程和确定性过程的相对重要性会受到环境变化因素持续时间的影响。Guo等(2018)对美国高草草原连续6年的增温研究发现, 与确定性过程相比, 随机性过程更能解释土壤生物分类学和进化发育组成; 但是随着增温年限的增加, 随机性过程的相对作用逐渐降低, 确定性的环境过滤作用的相对重要性逐渐增加。
3.2 生态过滤与进化适应的相对重要性
在整个大陆尺度上, 土壤pH值是影响细菌多样性的最重要的生态因子(Fierer & Jackson, 2006; Lauber et al, 2009; Rousk et al, 2010)。陆地生态系统起源于海洋(Nisbet & Sleep, 2001; Martin et al, 2008), 因此陆地生态系统的原始土壤偏弱碱性, 但长期的生物地球化学过程使得有些生态系统逐渐酸化(Wu, 1994; Fierer & Jackson, 2006)。现有研究多关注于长期土壤酸化影响细菌群落的生态过滤作用, 而忽略进化适应过程的潜在作用。理论上, 生物群落与环境之间的关系有两个方面: 一方面环境变化会通过生态过滤作用去除环境适应性低的物种, 从而降低群落多样性; 另一方面, 生物会通过进化过程提高其对环境的适应性, 或者适应环境的新物种从群落外部迁移过来, 从而提高群落多样性。因此, 有三种潜在的机制类型: (1)生态过滤和进化适应两种过程都不起作用; (2)仅仅生态过滤起作用; (3)进化适应抵消了生态过滤的部分作用效果。Zhang等(2015)提出一套理论分析方法, 通过比较野外长期酸化土壤样品与室内人工快速酸化实验样品的细菌多样性的差别探究生态过滤和进化适应作用的机制。他们认为室内人工快速酸化实验发生在较小的时间尺度上, 主要是生态过滤过程起作用; 野外长期土壤酸化发生在较大的时间尺度上, 生态过滤和进化适应共同起作用。通过推测两种过程在两种时间尺度上的7种组合情况及每种情况对应的多样性模式, 根据观察到的微生物多样性变化模式, 即可反推其作用机理。研究发现三种机制都存在, 但不同的微生物类群由不同类型的机制驱动, 第三种机制是最主要的作用类型。整个细菌域作为一个群体是由第三种机制驱动。Li等(2016)认为该研究对理解土壤酸化降低微生物多样性机制有一定启示意义。
3.3 中度干扰与资源生态位的共同作用
人类活动引起的环境变化导致了土壤微生物功能多样性的快速丧失(Liang et al, 2009; Paula et al, 2014; Singh et al, 2014; Zhang et al, 2017a)。但是, 我们对能够有效抵消或缓解多样性丧失的生态学过程和机制知之甚少。大量研究表明中度干扰和充分的有效资源量能够增加高等生物的多样性(Grime, 1973; Horn, 1975; Connell, 1978; Huston, 1994), 目前尚不清楚这些因素及其共同作用是否对微生物功能多样性的维持同样起作用。Zhang等(2018)研究了内蒙古典型草原中度干扰和有效资源量对土壤微生物群落多样性的影响机制。研究发现在所有具有中度干扰效果的处理中, 资源生态位维度与微生物基因丰富度之间有显著的正相关关系(r > 0.6, P < 0.01); 与之相反, 在其他低干扰处理中, 基因丰富度与资源生态位维度之间没有相关关系。总而言之, 在中度干扰和充足的资源生态位维度同时存在时, 微生物功能多样性最大。这意味着需要同时调控干扰强度和资源生态位才能有效地维持较高的土壤微生物多样性。
3.4 功能基因的核心成分和附属成分具有不同的驱动因子
生态学研究的一个核心问题是探究全球变化因子对土壤微生物群落的影响(Horz et al, 2004; Zhang et al, 2016b; Zhou et al, 2016)。微生物群落基因组中的不同基因具有不同的生态学重要性。Qin等(2010)认为微生物群落的功能基因分为核心成分和附属成分两部分: 核心成分包括单个微生物个体维持自身生长所必需的基因及微生物群落维持群落内稳态所必需的基因; 附属成分则指在特定情况下生存所需要的基因。微生物群落丰富度主要由附属成分的出现与否决定, 而微生物群落组成的变化则主要取决于核心成分相对丰度的变化(Fierer et al, 2012)。由于其不同的特征,群落功能基因中的核心成分和附属成分可能对不同的环境因子具有不同的响应(Zhang et al, 2019)。内蒙古草原的一个长达5年的氮素添加和增雨实验发现, 随着氮或土壤含水量的增加, 土壤微生物群落功能基因的核心成分和附属成分都会发生显著的变化, 但这些环境因子对功能基因的不同成分有不同的影响。氮添加通过氮循环过程影响微生物群落功能基因的核心成分, 而土壤含水量升高主要通过碳循环过程调控微生物群落功能基因的附属成分(Zhang et al, 2019)。目前关于微生物群落功能基因组不同组分的研究多集中于人类肠道菌群(Qin et al, 2010), 关于土壤微生物群落的较少。Burke等(2011)指出细菌群落主要以群落功能基因而非物种为基础进行群落构建。因此, 加强土壤微生物功能基因不同组分对环境变化的影响研究将为微生物群落的构建机制的研究提供新思路。
4 未来研究展望
目前研究者越来越关注全球变化对土壤微生物群落多样性的影响, 但迄今为止有关全球变化背景下微生物群落多样性的维持机制研究仍显不足。未来土壤微生物多样性研究应从加强全球变化多因素综合研究、微生物多样性维持的生态学机制研究、地上与地下多样性关联机制的研究、全球大尺度多生态系统的整合研究4个方面展开。
4.1 加强全球变化多因素综合研究
全球变化是一个多因素现象, 但目前的研究多从单因素角度研究环境变化对生态系统或土壤微生物群落的影响, 忽视各因素之间的耦合交互作用。在已发表的关于全球变化对土壤生物和过程的影响的研究中, 单因子实验高达80%, 19%涉及2种环境变化因素, 仅有不足2%的研究关注3个或多个环境因素(Rillig et al, 2019)。已有研究表明, 不同影响因素对微生物群落的影响相对大小不同(Zhang, 2016a), 且不同因素之间有着协同或拮抗等相互作用(Zhang et al, 2014b; Li et al, 2016)。之前的研究由于实验设计上的困难和经费的限制, 多因子实验不能同时考虑多个因素, 最多只考虑4个因素。参考生物多样性对生态系统功能的影响的研究方法, Rillig等(2019)发现, 尽管不同的环境变化因素对土壤理化性质、土壤过程和土壤微生物群落有着促进、抑制作用或无影响, 随着环境因子数的增加, 微生物群落特征确定性地向某一方向变化, 而与具体的环境因子组合无关。因此加强全球变化多因素对土壤微生物群落影响的研究, 有助于发现环境变化对微生物群落多样性影响的最一般的规律, 从而更准确地预测环境变化对微生物群落及其功能的影响, 进而预测整个生态系统对未来环境变化的响应。
4.2 加强微生物多样性维持的生态学机制的研究
生物多样性维持的理论机制是生态学的核心问题之一(徐冰和张大勇, 2014)。尽管生物学家和生态学家对动植物生物多样性维持机制已有了较为成熟的研究结果, 而对于微生物多样性维持机制尚待明确(Horner-Devine et al, 2004; Martiny et al, 2006)。目前大量研究多是从物理化学机制的角度研究环境变化对微生物群落多样性的影响, 但关于其内在的生态学机制还研究较少。在微生物生态学研究中, 目前大多是借鉴动植物的生态学理论(Martiny et al, 2006)。但因为微生物的生活史特征有别于动植物, 其多样性维持机制也会不同。加强微生物多样性维持的生态学机制的研究, 有利于检验和进一步发展生态学理论, 进而推动生态学研究的整体发展。
4.3 加强地上与地下多样性关联机制的研究
陆地生态系统由相互作用的地上部分和地下部分组成。地上和地下部分通过食物链、生物地球化学循环过程等紧密地联系在一起, 进而在多样性上产生关联。通过植物群落多样性可预测土壤微生物的α或β多样性(van der Heijden et al, 1998; Kowalchuk et al, 2002; Prober et al, 2015)。但目前关于环境变化对地上地下生物多样性关系影响的研究十分匮乏, 作用机制尚不明确。已有研究表明, 增雨会改变由氮添加引起的植物-微生物正向反馈关系(Li et al, 2016)。因此, 深入研究全球变化背景下地上-地下生物多样性之间的关系, 能更好地预测地上和地下生物多样性对未来全球变化的响应。
4.4 加强全球大尺度多生态系统的整合研究
环境变化对土壤微生物群落的影响与土壤初始情况、环境指标的相对改变量、地上植物群落组成等因素有关(孙良杰等, 2012; Feng et al, 2018; Wang et al, 2018), 因此全球变化对土壤微生物多样性的影响及其维持机制可能有生态系统依赖性。在全球范围内, 虽然氮添加总体上显著降低土壤微生物群落的多样性, 但它对土壤微生物群落多样性的影响具有生态系统依赖性。在草原、苔原、荒漠/灌丛生态系统中, 氮添加降低土壤微生物多样性, 但在森林生态系统中则增加其多样性(Wang et al, 2018)。在内蒙古草原, 氮添加主要通过改变土壤pH值进而影响土壤微生物群落, 但在土壤pH值本身就较低的森林生态系统中, 氮添加所引起的pH值降低是否起主要作用, 需要进一步研究。因此, 加强全球变化背景下大尺度多生态系统的整合研究有助于更准确地预测全球变化对生态系统的影响。
参考文献
Cryptic biodiversity loss linked to global climate change
Belowground biodiversity and ecosystem functioning
Soil fungal community structure in a temperate upland grassland soil
Bacterial community assembly based on functional genes rather than species
Watering increased DOC concentration but decreased N2O emission from a mixed grassland soil under different defoliation regimes
A preliminary theoretical framework of microbial ecology
微生物生态学理论框架
Stochastic and deterministic processes interact in the assembly of desert microbial communities on a global scale
DOI:10.1038/ismej.2011.21
URL
PMID:21368908
[本文引用: 1]

Extreme arid regions in the worlds' major deserts are typified by quartz pavement terrain. Cryptic hypolithic communities colonize the ventral surface of quartz rocks and this habitat is characterized by a relative lack of environmental and trophic complexity. Combined with readily identifiable major environmental stressors this provides a tractable model system for determining the relative role of stochastic and deterministic drivers in community assembly. Through analyzing an original, worldwide data set of 16S rRNA-gene defined bacterial communities from the most extreme deserts on the Earth, we show that functional assemblages within the communities were subject to different assembly influences. Null models applied to the photosynthetic assemblage revealed that stochastic processes exerted most effect on the assemblage, although the level of community dissimilarity varied between continents in a manner not always consistent with neutral models. The heterotrophic assemblages displayed signatures of niche processes across four continents, whereas in other cases they conformed to neutral predictions. Importantly, for continents where neutrality was either rejected or accepted, assembly drivers differed between the two functional groups. This study demonstrates that multi-trophic microbial systems may not be fully described by a single set of niche or neutral assembly rules and that stochasticity is likely a major determinant of such systems, with significant variation in the influence of these determinants on a global scale.
Stochastic community assembly causes higher biodiversity in more productive environments
Meta-analysis shows positive effects of plant diversity on microbial biomass and respiration
Abundance and community structure of ammonia- oxidizing archaea and bacteria in response to fertilization and mowing in a temperate steppe in Inner Mongolia
Nitrogen deposition and precipitation induced phylogenetic clustering of arbuscular mycorrhizal fungal communities
Six-year fertilization modifies the biodiversity of arbuscular mycorrhizal fungi in a temperate steppe in Inner Mongolia
Diversity in tropical rain forests and coral reefs
Estimating prokaryotic diversity and its limits
‘Everything is everywhere, but, the environment selects’; What did Baas Becking and Beijerinck really say?
Nitrogen and sulfur deposition on regional and global scales: A multimodel evaluation
Global state of biodiversity and loss
Long-term effects of imbalanced fertilization on the composition and diversity of soil bacterial community
Two key features influencing community assembly processes at regional scale: Initial state and degree of change in environmental conditions
The diversity and biogeography of soil bacterial communities
Cross-biome metagenomic analyses of soil microbial communities and their functional attributes
Transformation of the nitrogen cycle: Recent trends, questions, and potential solutions
Increased precipitation, rather than warming, exerts a strong influence on arbuscular mycorrhizal fungal community in a semiarid steppe ecosystem
A niche for ecosystem multifunctionality in global change research
Control of species density in herbaceous vegetation
Climate warming leads to divergent succession of grassland microbial communities
Beyond biogeographic patterns: Processes shaping the microbial landscape
Field experimental evidence that stochastic processes predominate in the initial assembly of bacterial communities
Progress in the studies of microbial diversity
微生物多样性研究进展与展望
Terrestrial ecosystem carbon dynamics and climate feedbacks
DOI:10.1038/nature06591 URL PMID:18202646 [本文引用: 1]
Interactions between aboveground and belowground biodiversity in terrestrial ecosystems: Patterns, mechanisms, and feedbacks
An ecological perspective on bacterial biodiversity
Ammonia-oxidizing bacteria respond to multifactorial global change
Soil microbiomes and climate change
Effects of simulated nitrogen deposition and precipitation manipulation on soil microorganisms in the desert steppe of Northern China
DOI:10.1590/18069657rbcs20180031 URL [本文引用: 1]
Temperature sensitivity of soil respiration rates enhanced by microbial community response
Arbuscular mycorrhizal fungal community response to warming and nitrogen addition in a semiarid steppe ecosystem
Effects of above-ground plant species composition and diversity on the diversity of soil-borne microorganisms
Pyrosequencing-based assessment soil pH as a predictor of soil bacterial community structure at the continental scale
DOI:10.1128/AEM.00335-09
URL
PMID:19502440
[本文引用: 1]
Soils harbor enormously diverse bacterial populations, and soil bacterial communities can vary greatly in composition across space. However, our understanding of the specific changes in soil bacterial community structure that occur across larger spatial scales is limited because most previous work has focused on either surveying a relatively small number of soils in detail or analyzing a larger number of soils with techniques that provide little detail about the phylogenetic structure of the bacterial communities. Here we used a bar-coded pyrosequencing technique to characterize bacterial communities in 88 soils from across North and South America, obtaining an average of 1,501 sequences per soil. We found that overall bacterial community composition, as measured by pairwise UniFrac distances, was significantly correlated with differences in soil pH (r = 0.79), largely driven by changes in the relative abundances of Acidobacteria, Actinobacteria, and Bacteroidetes across the range of soil pHs. In addition, soil pH explains a significant portion of the variability associated with observed changes in the phylogenetic structure within each dominant lineage. The overall phylogenetic diversity of the bacterial communities was also correlated with soil pH (R(2) = 0.50), with peak diversity in soils with near-neutral pHs. Together, these results suggest that the structure of soil bacterial communities is predictable, to some degree, across larger spatial scales, and the effect of soil pH on bacterial community composition is evident at even relatively coarse levels of taxonomic resolution.
Responses of soil bacterial communities to nitrogen deposition and precipitation increment are closely linked with aboveground community variation
DOI:10.1007/s00248-016-0730-z
URL
PMID:26838999
[本文引用: 5]
It has been predicted that precipitation and atmospheric nitrogen (N) deposition will increase in northern China; yet, ecosystem responses to the interactive effects of water and N remain largely unknown. In particular, responses of belowground microbial community to projected global change and their potential linkages to aboveground macro-organisms are rarely studied. In this study, we examined the responses of soil bacterial diversity and community composition to increased precipitation and multi-level N deposition in a temperate steppe in Inner Mongolia, China, and explored the diversity linkages between aboveground and belowground communities. It was observed that N addition caused the significant decrease in bacterial alpha-diversity and dramatic changes in community composition. In addition, we documented strong correlations of alpha- and beta-diversity between plant and bacterial communities in response to N addition. It was found that N enriched the so-called copiotrophic bacteria, but reduced the oligotrophic groups, primarily by increasing the soil inorganic N content and carbon availability and decreasing soil pH. We still highlighted that increased precipitation tended to alleviate the effects of N on bacterial diversity and dampen the plant-microbe connections induced by N. The counteractive effects of N addition and increased precipitation imply that even though the ecosystem diversity and function are predicted to be negatively affected by N deposition in the coming decades; the combination with increased precipitation may partially offset this detrimental effect.
Net ecosystem carbon dioxide exchange over grazed steppe in central Mongolia
Inner Mongolian steppe arbuscular mycorrhizal fungal communities respond more strongly to water availability than to nitrogen fertilization
Microarray-based analysis of microbial functional diversity along an oil contamination gradient in oil field
DOI:10.1111/j.1574-6941.2009.00774.x
URL
PMID:19780823
[本文引用: 1]
To understand better the in situ microbial functional diversity under oil contamination stress, soils were sampled along a contamination gradient at an oil field in north-east China. Microbial community functional structure was examined with a functional gene array, termed GeoChip. Multivariate statistical analysis and meta-analysis were conducted to study the functional gene responses to oil concentrations. The total functional gene abundance and diversity decreased along the gradient of increasing contamination. The overall abundance of soil bacteria, archaea and fungi decreased to 10%, 40% and 80% of those in the pristine soil. Several functional genes in the families pgl, rbcL, nifH and nor and those encoding cellulase, laccase, chitinase, urease and key enzymes in metabolizing organic compounds were significantly decreased with oil contamination, especially under high contamination stress. However, a few genes encoding key enzymes for catechol, protocatechuate, and biphenyl degradation and in the gene families of nir, rbcL and pgl showed a significant increase at a medium level of oil contamination. Oil content and soil available nitrogen were found to be important factors influencing the microbial community structure. The results provide an insight into microbial functional diversity in oil-contaminated soils, providing potential information for on-site management and remediation measures.
Differential responses of soil bacterial communities to long-term N and P inputs in a semi-arid steppe
Factors related to diversity of decomposer fungi in tropical forests
Metagenomic analysis of a permafrost microbial community reveals a rapid response to thaw
Hydrothermal vents and the origin of life
DOI:10.1038/nrmicro1991
URL
PMID:18820700
[本文引用: 1]
Submarine hydrothermal vents are geochemically reactive habitats that harbour rich microbial communities. There are striking parallels between the chemistry of the H(2)-CO(2) redox couple that is present in hydrothermal systems and the core energy metabolic reactions of some modern prokaryotic autotrophs. The biochemistry of these autotrophs might, in turn, harbour clues about the kinds of reactions that initiated the chemistry of life. Hydrothermal vents thus unite microbiology and geology to breathe new life into research into one of biology's most important questions - what is the origin of life?
Microbial biogeography: Putting microorganisms on the map
Methane dynamics regulated by microbial community response to permafrost thaw
The habitat and nature of early life
DOI:10.1038/35059210
URL
PMID:11234022
[本文引用: 1]
Earth is over 4,500 million years old. Massive bombardment of the planet took place for the first 500-700 million years, and the largest impacts would have been capable of sterilizing the planet. Probably until 4,000 million years ago or later, occasional impacts might have heated the ocean over 100 degrees C. Life on Earth dates from before about 3,800 million years ago, and is likely to have gone through one or more hot-ocean 'bottlenecks'. Only hyperthermophiles (organisms optimally living in water at 80-110 degrees C) would have survived. It is possible that early life diversified near hydrothermal vents, but hypotheses that life first occupied other pre-bottleneck habitats are tenable (including transfer from Mars on ejecta from impacts there). Early hyperthermophile life, probably near hydrothermal systems, may have been non-photosynthetic, and many housekeeping proteins and biochemical processes may have an original hydrothermal heritage. The development of anoxygenic and then oxygenic photosynthesis would have allowed life to escape the hydrothermal setting. By about 3,500 million years ago, most of the principal biochemical pathways that sustain the modern biosphere had evolved, and were global in scope.
Land use change alters functional gene diversity, composition and abundance in Amazon forest soil microbial communities
DOI:10.1111/mec.12786
URL
PMID:24806276
[本文引用: 1]
Land use change in the Amazon rainforest alters the taxonomic structure of soil microbial communities, but whether it alters their functional gene composition is unknown. We used the highly parallel microarray technology GeoChip 4.0, which contains 83,992 probes specific for genes linked nutrient cycling and other processes, to evaluate how the diversity, abundance and similarity of the targeted genes responded to forest-to-pasture conversion. We also evaluated whether these parameters were reestablished with secondary forest growth. A spatially nested scheme was employed to sample a primary forest, two pastures (6 and 38 years old) and a secondary forest. Both pastures had significantly lower microbial functional genes richness and diversity when compared to the primary forest. Gene composition and turnover were also significantly modified with land use change. Edaphic traits associated with soil acidity, iron availability, soil texture and organic matter concentration were correlated with these gene changes. Although primary and secondary forests showed similar functional gene richness and diversity, there were differences in gene composition and turnover, suggesting that community recovery was not complete in the secondary forest. Gene association analysis revealed that response to ecosystem conversion varied significantly across functional gene groups, with genes linked to carbon and nitrogen cycling mostly altered. This study indicates that diversity and abundance of numerous environmentally important genes respond to forest-to-pasture conversion and hence have the potential to affect the related processes at an ecosystem scale.
Plant diversity predicts beta but not alpha diversity of soil microbes across grasslands worldwide
A human gut microbial gene catalogue established by metagenomic sequencing
DOI:10.1038/nature08821
URL
PMID:20203603
[本文引用: 2]
To understand the impact of gut microbes on human health and well-being it is crucial to assess their genetic potential. Here we describe the Illumina-based metagenomic sequencing, assembly and characterization of 3.3 million non-redundant microbial genes, derived from 576.7 gigabases of sequence, from faecal samples of 124 European individuals. The gene set, approximately 150 times larger than the human gene complement, contains an overwhelming majority of the prevalent (more frequent) microbial genes of the cohort and probably includes a large proportion of the prevalent human intestinal microbial genes. The genes are largely shared among individuals of the cohort. Over 99% of the genes are bacterial, indicating that the entire cohort harbours between 1,000 and 1,150 prevalent bacterial species and each individual at least 160 such species, which are also largely shared. We define and describe the minimal gut metagenome and the minimal gut bacterial genome in terms of functions present in all individuals and most bacteria, respectively.
Responses of soil total microbial biomass and community compositions to rainfall reductions
The role of multiple global change factors in driving soil functions and microbial biodiversity
Soil bacterial and fungal communities across a pH gradient in an arable soil
DOI:10.1038/ismej.2010.58
URL
PMID:20445636
[本文引用: 1]
Soils collected across a long-term liming experiment (pH 4.0-8.3), in which variation in factors other than pH have been minimized, were used to investigate the direct influence of pH on the abundance and composition of the two major soil microbial taxa, fungi and bacteria. We hypothesized that bacterial communities would be more strongly influenced by pH than fungal communities. To determine the relative abundance of bacteria and fungi, we used quantitative PCR (qPCR), and to analyze the composition and diversity of the bacterial and fungal communities, we used a bar-coded pyrosequencing technique. Both the relative abundance and diversity of bacteria were positively related to pH, the latter nearly doubling between pH 4 and 8. In contrast, the relative abundance of fungi was unaffected by pH and fungal diversity was only weakly related with pH. The composition of the bacterial communities was closely defined by soil pH; there was as much variability in bacterial community composition across the 180-m distance of this liming experiment as across soils collected from a wide range of biomes in North and South America, emphasizing the dominance of pH in structuring bacterial communities. The apparent direct influence of pH on bacterial community composition is probably due to the narrow pH ranges for optimal growth of bacteria. Fungal community composition was less strongly affected by pH, which is consistent with pure culture studies, demonstrating that fungi generally exhibit wider pH ranges for optimal growth.
Spatial scale affects the relative role of stochasticity versus determinism in soil bacterial communities in wheat fields across the North China Plain
Loss of microbial diversity in soils is coincident with reductions in some specialized functions
DOI:10.1111/1462-2920.12353
URL
PMID:24422656
[本文引用: 1]
Loss of microbial diversity is considered a major threat because of its importance for ecosystem functions, but there is a lack of conclusive evidence that diversity itself is reduced under anthropogenic stress, and about the consequences of diversity loss. Heavy metals are one of the largest, widespread pollutant types globally, and these represent a significant environmental stressor for terrestrial microbial communities. Using combined metagenomics and functional assays, we show that the compositional and functional response of microbial communities to long-term heavy metal stress results in a significant loss of diversity. Our results indicate that even at a moderate loss of diversity, some key specialized functions (carried out by specific groups) may be compromised. Together with previous work, our data suggest disproportionate impact of contamination on microbes that carry out specialized, but essential, ecosystem functions. Based on these findings, we propose a conceptual framework to explicitly consider diversity of functions and microbial functional groups to test the relationship between biodiversity and soil functions.
Research progress of the effect of global change on soil microbial diversity in the grassland
全球变化对草地土壤微生物群落多样性的影响研究进展
Trophic regulations of the soil microbiome
DOI:10.1016/j.tim.2019.04.008
URL
PMID:31138481
[本文引用: 1]
The soil microbiome regulates vital ecosystem functions ranging from primary production to soil carbon sequestration. Yet, we have only begun to understand the factors regulating the soil microbiome. While the importance of abiotic factors is increasingly recognized, the roles of trophic regulations in driving the structure and function of the soil microbiome remain less explored. Here, we review the current understanding of how and when microbial and top predators of the soil shape the community structure and function of the soil microbiome via both direct and indirect effects. We finally highlight that the structure and function of the soil microbiome depend on the interactive effects among predation, plant inputs, and abiotic variables present in the soil.
Productivity and sustainability influenced by biodiversity in grassland ecosystems
Mycorrhizal fungal diversity determines plant biodiversity, ecosystem variability and productivity
Bacterial targets as potential indicators of diesel fuel toxicity in subantartic soils
Resource availability controls fungal diversity across a plant diversity gradient
DOI:10.1111/j.1461-0248.2006.00965.x
URL
PMID:16972876
[本文引用: 1]
Despite decades of research, the ecological determinants of microbial diversity remain poorly understood. Here, we test two alternative hypotheses concerning the factors regulating fungal diversity in soil. The first states that higher levels of plant detritus production increase the supply of limiting resources (i.e. organic substrates) thereby increasing fungal diversity. Alternatively, greater plant diversity increases the range of organic substrates entering soil, thereby increasing the number of niches to be filled by a greater array of heterotrophic fungi. These two hypotheses were simultaneously examined in experimental plant communities consisting of one to 16 species that have been maintained for a decade. We used ribosomal intergenic spacer analysis (RISA), in combination with cloning and sequencing, to quantify fungal community composition and diversity within the experimental plant communities. We used soil microbial biomass as a temporally integrated measure of resource supply. Plant diversity was unrelated to fungal diversity, but fungal diversity was a unimodal function of resource supply. Canonical correspondence analysis (CCA) indicated that plant diversity showed a relationship to fungal community composition, although the occurrence of RISA bands and operational taxonomic units (OTUs) did not differ among the treatments. The relationship between fungal diversity and resource availability parallels similar relationships reported for grasslands, tropical forests, coral reefs, and other biotic communities, strongly suggesting that the same underlying mechanisms determine the diversity of organisms at multiple scales.
Decreasing soil microbial diversity is associated with decreasing microbial biomass under nitrogen addition
DOI:10.1016/j.soilbio.2018.02.003 URL [本文引用: 4]
Grassland ecology in China: Perspectives and challenges
Habitat-specific patterns and drivers of bacterial beta-diversity in China's drylands
DOI:10.1038/ismej.2017.11
URL
PMID:28282041
[本文引用: 2]
The existence of biogeographic patterns among most free-living microbial taxa has been well established, yet little is known about the underlying mechanisms that shape these patterns. Here, we examined soil bacterial beta-diversity across different habitats in the drylands of northern China. We evaluated the relative importance of environmental factors versus geographic distance to a distance-decay relationship, which would be explained by the relative effect of basic ecological processes recognized as drivers of diversity patterns in macrobial theoretical models such as selection and dispersal. Although the similarity of bacterial communities significantly declined with increasing geographic distance, the distance-decay slope and the relative importance of factors driving distance-decay patterns varied across different habitats. A strong distance-decay relationship was observed in the alpine grassland, where the community similarity was influenced only by the environmental factors. In contrast, geographic distance was solely responsible for community similarity in the desert. Even the average compositional similarity among locations in the desert was distinctly lower compared with those in other habitats. We found no evidence that dispersal limitation strongly influenced the beta-diversity of bacterial communities in the desert grassland and typical grassland. Together, our results provide robust evidence of habitat specificity for microbial diversity patterns and their underlying drivers. Our findings suggest that microorganisms also have multiple drivers of diversity patterns and some of which may be parallel to some fundamental processes for explaining biodiversity patterns in macroorganisms.
Progress on the biodiversity of microorganisms: Patterns and processes
微生物多样性及其分布的研究进展: 模式与过程
Long-term inorganic fertilizer use influences bacterial communities in Mollisols of Northeast China based on high-throughput sequencing and network analyses
Impacts of long-term nitrogen addition, watering and mowing on ammonia oxidizers, denitrifiers and plant communities in a temperate steppe
Nitrogen deposition combined with elevated precipitation is conducive to maintaining the stability of the soil fungal diversity on the Stipa baicalensis steppe
Nitrogen deposition mediates the effects and importance of chance in changing biodiversity
DOI:10.1111/j.1365-294X.2010.04933.x
URL
PMID:21087326
[本文引用: 2]
Nitrogen deposition is changing biodiversity on Earth. We need to understand the underlying mechanisms to conserve biodiversity better. Both selection and chance are potential mechanisms, and they may operate concurrently. Then, what are the respective effects of selection and chance, what is their relative importance and how do they change with increasing nitrogen deposition rate? Here, we performed a 6-year nitrogen addition experiment (0-28 g N/m(2) /year) in a typical steppe ecosystem of Inner Mongolia to investigate the community structure of plants, bacteria and ammonia-oxidizing Archaea (AOA). We developed an experimentally based calculation method to first separate the structural variations between plots into the effects of selection (S) and chance (C), and then calculate their relative importance. Our results showed that as nitrogen addition rate increased, S for both plants and bacteria increased, but C for plants first increased and then decreased, and C for bacteria also increased; meanwhile, both S and C for AOA changed nonlinearly. As nitrogen addition rate increased, the importance of chance decreased on the whole for all these communities, but it decreased nonlinearly for plants and bacteria, with a local increase at certain intermediate rates. At all treatments, the importance of chance was <0.5 for plants, but >0.5 for AOA. These results demonstrated that nitrogen deposition changed biodiversity by mediating the effects and importance of chance, implicating different strategies should be adopted in conserving biodiversity according to nitrogen deposition rate and community properties.
Nitrogen deposition alters soil chemical properties and bacterial communities in the Inner Mongolia grassland
Soil bacterial communities respond to mowing and nutrient addition in a steppe ecosystem
DOI:10.1371/journal.pone.0084210
URL
PMID:24391915
[本文引用: 1]
In many grassland ecosystems, nitrogen (N) and phosphorus (P) are added to improve plant productivity, and the aboveground plant biomass is mowed and stored as hay for the bullamacow. Nutrient addition and mowing affect the biodiversity and ecosystem functioning, and most of the previous studies have primarily focused on their effects on macro-organisms, neglecting the responses of soil microbial communities. In this study, we examined the changes in three community attributes (abundance, richness, and composition) of the entire bacterial kingdom and 16 dominant bacterial phyla/classes in response to mowing, N addition, P addition, and their combinations, by conducting a 5-year experiment in a steppe ecosystem in Inner Mongolia, China. Overall, N addition had a greater effect than mowing and P addition on most of these bacterial groups, as indicated by changes in the abundance, richness and composition in response to these treatments. N addition affected these soil bacterial groups primarily through reducing soil pH and increasing available N content. Meanwhile, the 16 bacterial phyla/classes responded differentially to these experimental treatments, with Acidobacteria, Acidimicrobidae, Deltaproteobacteria, and Gammaproteobacteria being the most sensitive. The changes in the abundance, richness, and composition of various bacterial groups could imply some potential shift in their ecosystem functions. Furthermore, the important role of decreased soil pH caused by N addition in affecting soil bacterial communities suggests the importance of restoring acidified soil to maintain soil bacterial diversity.
Response of the abundance of key soil microbial nitrogen-cycling genes to multi-factorial global changes
DOI:10.1371/journal.pone.0076500
URL
PMID:24124568
[本文引用: 1]
Multiple co-occurring environmental changes are affecting soil nitrogen cycling processes, which are mainly mediated by microbes. While it is likely that various nitrogen-cycling functional groups will respond differently to such environmental changes, very little is known about their relative responsiveness. Here we conducted four long-term experiments in a steppe ecosystem by removing plant functional groups, mowing, adding nitrogen, adding phosphorus, watering, warming, and manipulating some of their combinations. We quantified the abundance of seven nitrogen-cycling genes, including those for fixation (nifH), mineralization (chiA), nitrification (amoA of ammonia-oxidizing bacteria (AOB) or archaea (AOA)), and denitrification (nirS, nirK and nosZ). First, for each gene, we compared its sensitivities to different environmental changes and found that the abundances of various genes were sensitive to distinct and different factors. Overall, the abundances of nearly all genes were sensitive to nitrogen enrichment. In addition, the abundances of the chiA and nosZ genes were sensitive to plant functional group removal, the AOB-amoA gene abundance to phosphorus enrichment when nitrogen was added simultaneously, and the nirS and nirK gene abundances responded to watering. Second, for each single- or multi-factorial environmental change, we compared the sensitivities of the abundances of different genes and found that different environmental changes primarily affected different gene abundances. Overall, AOB-amoA gene abundance was most responsive, followed by the two denitrifying genes nosZ and nirS, while the other genes were less sensitive. These results provide, for the first time, systematic insights into how the abundance of each type of nitrogen-cycling gene and the equilibrium state of all these nitrogen-cycling gene abundances would shift under each single- or multi-factorial global change.
Soil bacterial communities respond to climate changes in a temperate steppe
DOI:10.1371/journal.pone.0078616
URL
PMID:24250803
[本文引用: 2]
Climate warming and shifting precipitation regimes are affecting biodiversity and ecosystem functioning. Most studies have focused on the influence of warming and altered precipitation on macro-organisms, whereas the responses of soil microbial communities have been neglected. We studied the changes in the abundance, richness, and composition of the entire bacterial kingdom and 16 dominant bacterial phyla/classes in response to increased precipitation, warming, and their combination, by conducting a 5-year experiment in a steppe ecosystem in Inner Mongolia, China. Watering had a greater effect than warming on almost all the bacterial groups as indicated by changes in all the three attributes (abundance, richness, and composition). The 16 phyla/classes responded differentially to the experimental treatments, with Acidobacteria and Gamma-proteobacteria being the most sensitive. Stepwise regression analyses further revealed that climate changes altered the abundance and richness of bacterial groups primarily through direct routes (e.g., increasing soil water content), and changed the community composition through both direct and indirect routes (e.g., reducing soil total nitrogen content and increasing soil pH). The diverse responses of various bacterial groups could imply some potential shift in their ecosystem functions under climate changes; meanwhile, the indirect routes that are important in altering bacterial composition suggest that specific strategies (e.g., adding NH4NO3 to maintain soil nitrogen content and pH) could be adopted to maintain soil microbial composition under climate changes.
Water content differences have stronger effects than plant functional groups on soil bacteria in a steppe ecosystem
DOI:10.1371/journal.pone.0115798
URL
PMID:25546333
[本文引用: 1]
Many investigations across natural and artificial plant diversity gradients have reported that both soil physicochemical factors and plant community composition affect soil microbial communities. To test the effect of plant diversity loss on soil bacterial communities, we conducted a five-year plant functional group removal experiment in a steppe ecosystem in Inner Mongolia (China). We found that the number and composition type of plant functional groups had no effect on bacterial diversity and community composition, or on the relative abundance of major taxa. In contrast, bacterial community patterns were significantly structured by soil water content differences among plots. Our results support researches that suggest that water availability is the key factor structuring soil bacterial communities in this semi-arid ecosystem.
The counteractive effects of nitrogen addition and watering on soil bacterial communities in a steppe ecosystem
Mechanisms of soil acidification reducing bacterial diversity
Environmental changes affect the assembly of soil bacterial community primarily by mediating stochastic processes
Stochastic processes play more important roles in driving the dynamics of rarer species
Decreased plant productivity resulting from plant group removal experiment constrains soil microbial functional diversity
DOI:10.1111/gcb.13783
URL
PMID:28585356
[本文引用: 3]
Anthropogenic environmental changes are accelerating the rate of biodiversity loss on Earth. Plant diversity loss is predicted to reduce soil microbial diversity primarily due to the decreased variety of carbon/energy resources. However, this intuitive hypothesis is supported by sparse empirical evidence, and most underlying mechanisms remain underexplored or obscure altogether. We constructed four diversity gradients (0-3) in a five-year plant functional group removal experiment in a steppe ecosystem in Inner Mongolia, China, and quantified microbial taxonomic and functional diversity with shotgun metagenome sequencing. The treatments had little effect on microbial taxonomic diversity, but were found to decrease functional gene diversity. However, the observed decrease in functional gene diversity was more attributable to a loss in plant productivity, rather than to the loss of any individual plant functional group per se. Reduced productivity limited fresh plant resources supplied to microorganisms, and thus, intensified the pressure of ecological filtering, favoring genes responsible for energy production/conversion, material transport/metabolism and amino acid recycling, and accordingly disfavored many genes with other functions. Furthermore, microbial respiration was correlated with the variation in functional composition but not taxonomic composition. Overall, the amount of carbon/energy resources driving microbial gene diversity was identified to be the critical linkage between above- and belowground communities, contrary to the traditional framework of linking plant clade/taxonomic diversity to microbial taxonomic diversity.
Experimental warming reveals positive feedbacks to climate change in the Eurasian Steppe
Effect of intermediate disturbance on soil microbial functional diversity depends on the amount of effective resources
DOI:10.1111/1462-2920.14407
URL
PMID:30209865
[本文引用: 1]
Many anthropogenic environmental changes are leading to a rapid decline in soil microbial functional diversity. However, ecological mechanisms that can serve to counteract/resist the diversity loss remain largely underexplored. In particular, although intermediate disturbance and increased amount of effective resources can promote the diversity of higher organisms, the potential role of these factors, and their combination, in maintaining microbial functional diversity is poorly studied. We conducted a 5-year experiment in a Eurasian steppe, manipulating mowing, nitrogen addition, phosphorus addition and their combinations. Nitrogen addition decreased soil pH by ~0.6 and bacterial abundance by ~19.5%, causing a disturbance effect. Phosphorus addition significantly decreased the effective amount of soil carbon-, nitrogen-, phosphorus- and water-relevant resources. Across all nitrogen-addition treatments subject to intermediate disturbance, there was a significant positive correlation between soil effective resource amount and microbial gene richness (r > 0.6, p < 0.01), which was elevated, in part, due to the increased fungal abundance. In contrast, significant correlations between gene richness and resource amount were not found under low-disturbance conditions. Overall, gene richness was greatest under conditions of both intermediate disturbance and ample effective resources, suggesting that the two factors could be manipulated in combination for the maintenance of microbial functional diversity.
Distinct drivers of core and accessory components of soil microbial community functional diversity under environmental changes
DOI:10.1128/mSystems.00374-19
URL
PMID:31575666
[本文引用: 2]
It is a central ecological goal to explore the effects of global change factors on soil microbial communities. The vast functional gene repertoire of soil microbial communities is composed of both core and accessory genes, which may be governed by distinct drivers. This intuitive hypothesis, however, remains largely unexplored. We conducted a 5-year nitrogen and water addition experiment in the Eurasian steppe and quantified microbial gene diversity via shotgun metagenomics. Nitrogen addition led to an 11-fold increase in the abundance (based on quantitative PCR [qPCR]) of ammonia-oxidizing bacteria, which have mainly core community genes and few accessory community genes. Thus, nitrogen addition substantially increased the relative abundance of many core genes at the whole-community level. Water addition stimulated both plant diversity and microbial respiration; however, increased carbon/energy resources from plants did not counteract increased respiration, so soil carbon/energy resources became more limited. Thus, water addition selected for microorganisms with genes responsible for degrading recalcitrant soil organic matter. Accordingly, many other microorganisms without these genes (but likely with other accessory community genes due to relatively stable average microbial genome size) were selected against, leading to the decrease in the diversity of accessory community genes. In summary, nitrogen addition primarily affected core community genes through nitrogen-cycling processes, and water addition primarily regulated accessory community genes through carbon-cycling processes. Although both gene components may significantly respond as the intensity of nitrogen/water addition increases, our results demonstrated how these common global change factors distinctly impact each component.IMPORTANCE Our results demonstrated increased ecosystem nitrogen and water content as the primary drivers of the core and accessory components of soil microbial community functional diversity, respectively. Our findings suggested that more attention should be paid to certain components of community functional diversity under specific global change conditions. Our findings also indicated that microbial communities have adapted to nitrogen addition by strengthening the function of ammonia oxidization to deplete the excess nitrogen, thus maintaining ecosystem homeostasis. Because community gene richness is primarily determined by the presence/absence of accessory community genes, our findings further implied that strategies such as maintaining the amount of soil organic matter could be adopted to effectively improve the functional gene diversity of soil microbial communities subject to global change factors.
Temperature mediates continental-scale diversity of microbes in forest soils
DOI:10.1038/ncomms12083
URL
PMID:27377774
[本文引用: 1]
Climate warming is increasingly leading to marked changes in plant and animal biodiversity, but it remains unclear how temperatures affect microbial biodiversity, particularly in terrestrial soils. Here we show that, in accordance with metabolic theory of ecology, taxonomic and phylogenetic diversity of soil bacteria, fungi and nitrogen fixers are all better predicted by variation in environmental temperature than pH. However, the rates of diversity turnover across the global temperature gradients are substantially lower than those recorded for trees and animals, suggesting that the diversity of plant, animal and soil microbial communities show differential responses to climate change. To the best of our knowledge, this is the first study demonstrating that the diversity of different microbial groups has significantly lower rates of turnover across temperature gradients than other major taxa, which has important implications for assessing the effects of human-caused changes in climate, land use and other factors.
Microbial mediation of carbon-cycle feedbacks to climate warming
Patterns and mechanisms of responses by soil microbial communities to nitrogen addition

{kind=link}
{kind=link}