
生物多样性 ›› 2017, Vol. 25 ›› Issue (6): 675-682. DOI: 10.17520/biods.2017042 cstr: 32101.14.biods.2017042
所属专题: 物种形成与系统进化
舒江平1,2, 刘莉1,2, 沈慧2, 戴锡玲1, 王全喜1,4, 严岳鸿2,3,4,*( )
)
收稿日期:2017-02-18
接受日期:2017-06-20
出版日期:2017-06-20
发布日期:2017-07-10
通讯作者:
严岳鸿
基金资助:
Jiangping Shu1,2, Li Liu1,2, Hui Shen2, Xiling Dai1, Quanxi Wang1,4, Yuehong Yan2,3,4,*()
Received:2017-02-18
Accepted:2017-06-20
Online:2017-06-20
Published:2017-07-10
Contact:
Yan Yuehong
摘要:
植物由水生走向陆生的进化过程中经历了非常复杂的演化, 期间产生的大量基因的进化路线可能互不相同, 因此仅仅使用系统发育树无法呈现真实的演化关系。系统发育网络图能够清楚地展示包括垂直演化和水平演化在内的复杂网状进化关系。本文选取莱茵衣藻(Chlamydomonas reinhardtii)和4种陆生植物, 利用系统基因组学的方法, 筛选得到1,668个一对一直系同源基因, 重新构建了陆生植物的系统发育网状进化关系。结果发现, 使用不同的分析策略所得到的系统发育树不同; 对1,668个基因单独分析, 发现存在15种不同的拓扑结构; 对5个物种筛选得到的直系同源基因进行系统发育网络分析显示, 在非常稳健的系统发育网络图中, 仅仅5个物种就存在9个不同的分离支, 暗示着非常复杂的网状进化关系; 而且藻类植物与苔藓植物和石松类植物的分离支之间差异很小, 这可能是产生系统发育树冲突的原因之一, 也暗示着早期陆生植物发生了复杂的辐射演化。
舒江平, 刘莉, 沈慧, 戴锡玲, 王全喜, 严岳鸿 (2017) 基于系统基因组学分析揭示早期陆生植物的复杂网状进化关系. 生物多样性, 25, 675-682. DOI: 10.17520/biods.2017042.
Jiangping Shu, Li Liu, Hui Shen, Xiling Dai, Quanxi Wang, Yuehong Yan (2017) The complex reticulate evolutionary relationships of early terrestrial plants as revealed by phylogenomics analysis. Biodiversity Science, 25, 675-682. DOI: 10.17520/biods.2017042.
| 物种 Species | 分类 Classification | BUSCO评估结果 BUSCO results |
|---|---|---|
| 福建观音座莲 Angiopteris fokiensis | 真蕨类植物 Monilophytes | C: 66.4% [S: 43.2%, D: 23.2%], F: 5.9%, M: 27.7%, n: 1440 |
| 欧洲云杉 Picea abies | 种子植物 Spermatophytes | C: 34.0% [S: 28.9%, D: 5.1%], F: 7.4%, M: 58.6%, n: 1,440 |
| 江南卷柏 Selaginella moellendorffii | 石松类植物 Lycophytes | C: 63.2% [S: 10.0%, D: 53.2%], F: 4.7%, M: 32.1%, n: 1,440 |
| 小立碗藓 Physcomitrella patens | 苔藓植物 Bryophytes | C: 70.1% [S: 46.0%, D: 24.1%], F: 2.6%, M: 27.3%, n: 1,440 |
| 莱茵衣藻 Chlamydomonas reintmrdtii | 藻类植物 Thallophytes | C: 18.8% [S: 17.9%, D: 0.9%], F: 1.7%, M: 79.5%, n: 1,440 |
表1 转录组和基因组组装完整性评估结果统计
Table 1 The assessment results of assembly completeness of transcriptome and genome
| 物种 Species | 分类 Classification | BUSCO评估结果 BUSCO results |
|---|---|---|
| 福建观音座莲 Angiopteris fokiensis | 真蕨类植物 Monilophytes | C: 66.4% [S: 43.2%, D: 23.2%], F: 5.9%, M: 27.7%, n: 1440 |
| 欧洲云杉 Picea abies | 种子植物 Spermatophytes | C: 34.0% [S: 28.9%, D: 5.1%], F: 7.4%, M: 58.6%, n: 1,440 |
| 江南卷柏 Selaginella moellendorffii | 石松类植物 Lycophytes | C: 63.2% [S: 10.0%, D: 53.2%], F: 4.7%, M: 32.1%, n: 1,440 |
| 小立碗藓 Physcomitrella patens | 苔藓植物 Bryophytes | C: 70.1% [S: 46.0%, D: 24.1%], F: 2.6%, M: 27.3%, n: 1,440 |
| 莱茵衣藻 Chlamydomonas reintmrdtii | 藻类植物 Thallophytes | C: 18.8% [S: 17.9%, D: 0.9%], F: 1.7%, M: 79.5%, n: 1,440 |
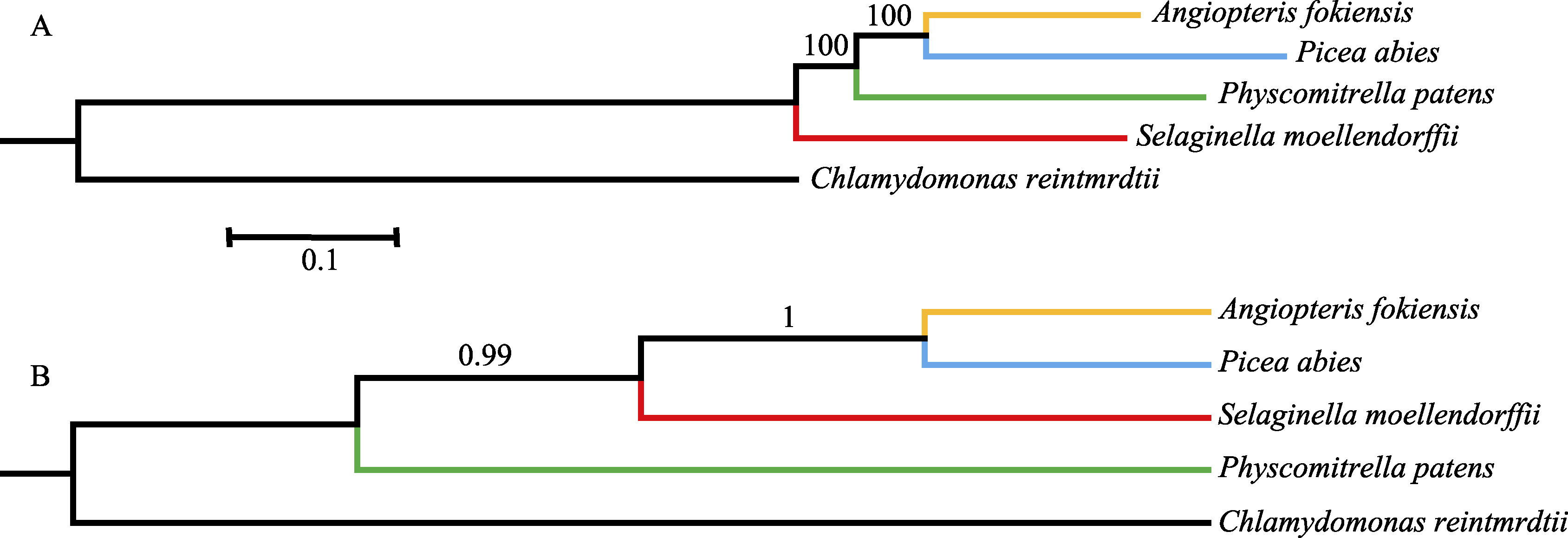
图1 基于串联和联合的方法分析得到的系统发育树。(A)使用串联矩阵构建的最大似然树; (B)使用联合基因树构建的物种树。
Fig. 1 The phylogenetic trees based on concatenation and coalescence methods. (A) The maximum likelihood tree based on concatenation method; (B) The species tree based on coalescence method.
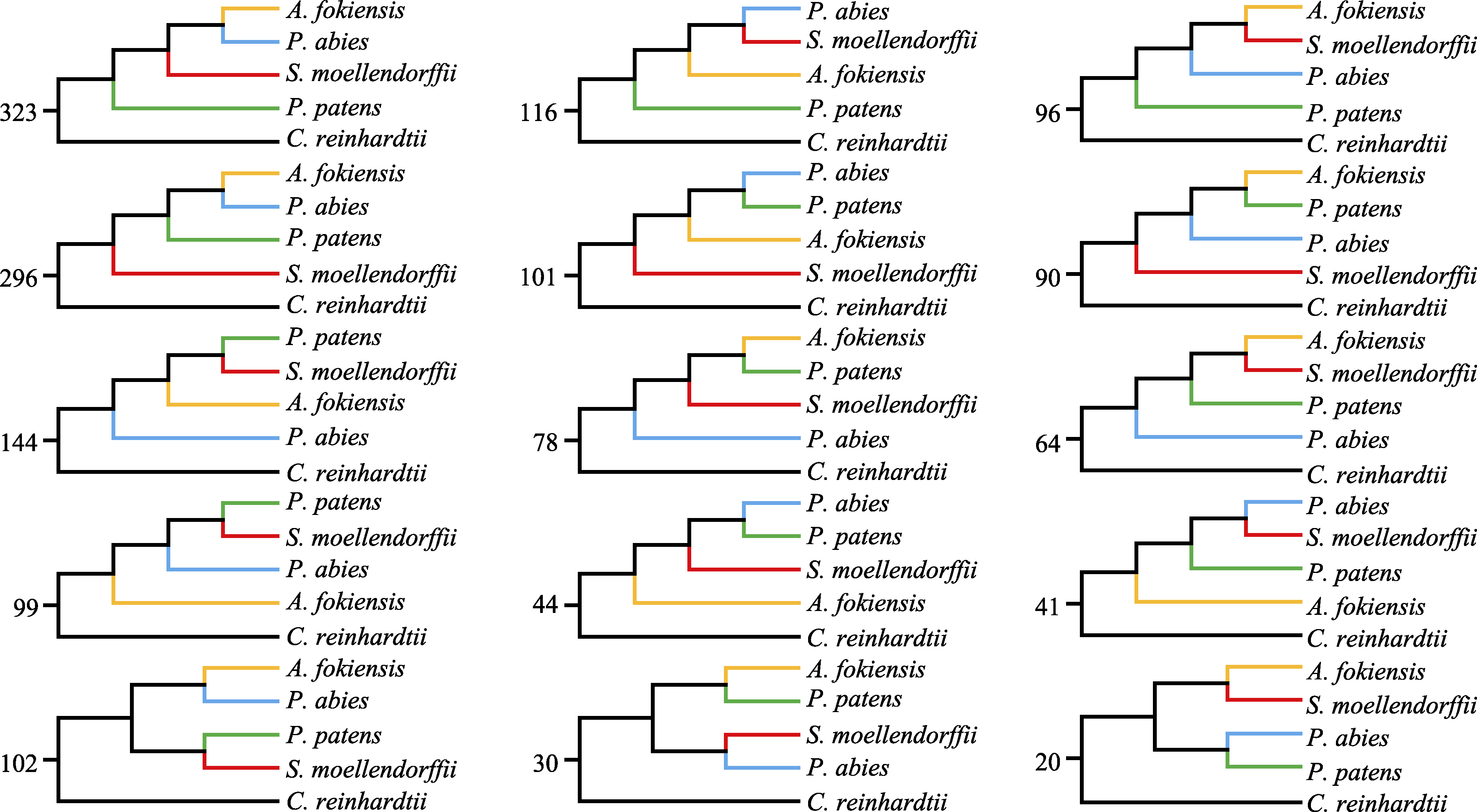
图2 使用最大似然法构建的15种拓扑结构的基因树。数字表示每种拓扑结构的数量。
Fig. 2 Fifteen topological structure of gene trees based on maximum likelihood method. The numbers mean the amount of the topological structure.
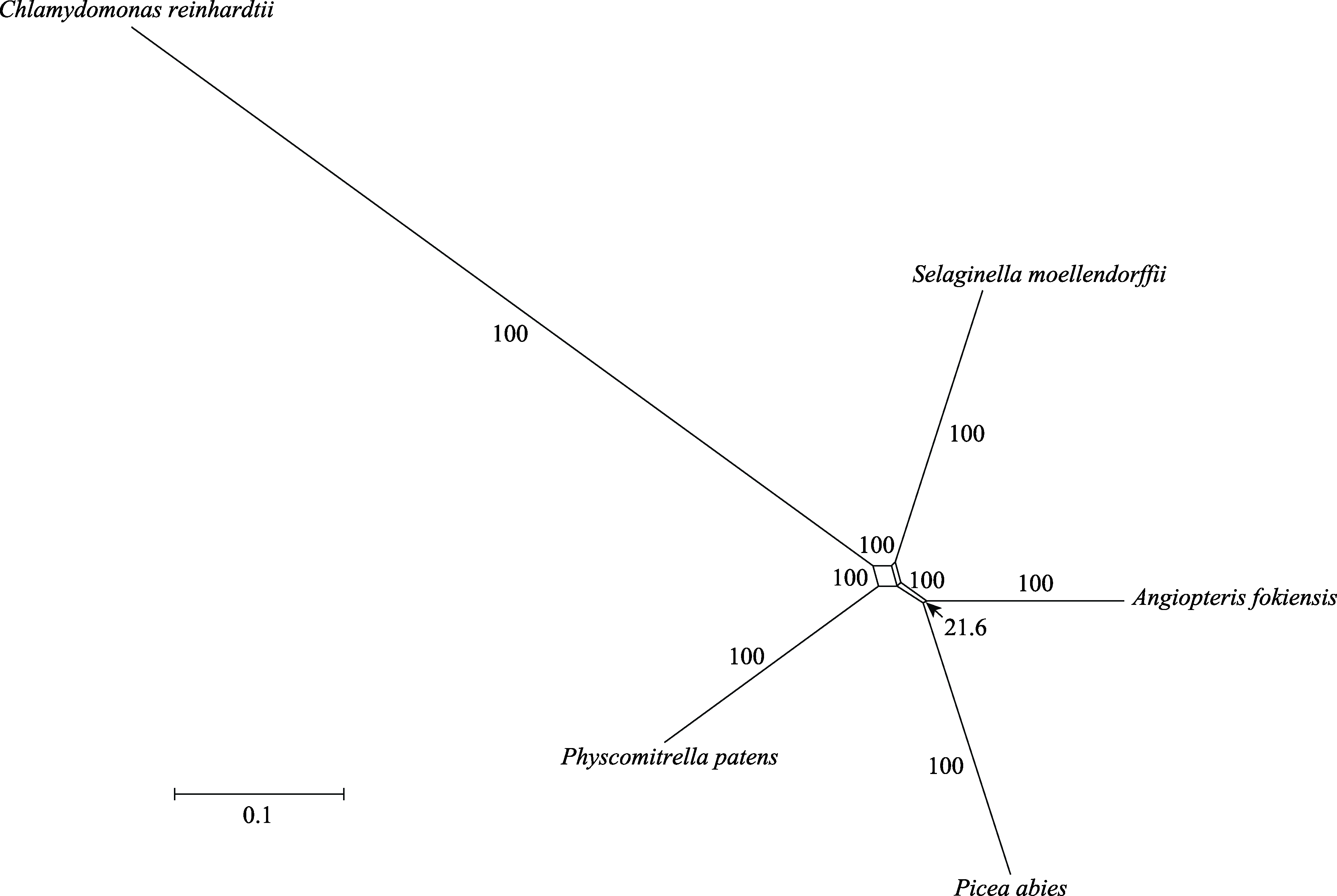
图3 基于1,668个基因构建的早期陆生植物的系统发育网络。数字表示每个分离支的支持率, 除了最短分离支(箭头)之外, 其他分离支的支持率都为100%; 平行的分离支为同一种分离支。
Fig. 3 The phylogenetic network of early land plants based on 1,668 genes. The numbers mean the bootstrap support of each split branch. In addition to the shortest split branch (arrow), the bootstrap support of other split branches is 100%. The parallel split branches are the same type of split branch.
| [1] | Arnold CA (2013) An Introduction to Paleobotany. Read Books Ltd., New York. |
| [2] | Bandelt HJ, Forster P, Röhl A (1999) Median-joining networks for inferring intraspecific phylogenies. Molecular Biology and Evolution, 16, 37-48. |
| [3] | Bohlin K (1901) Utkast till de gröna algernas och arkegoniaternas fylogeni. Almqvist & Wiksells Boktryckeri AB, Uppsala. |
| [4] | Bower FO (1935) Primitive land plants. Science, 81, 537-539. |
| [5] | Bryant D, Moulton V (2004) Neighbor-net: an agglomerative method for the construction of phylogenetic networks. Molecular Biology and Evolution, 21, 255-265. |
| [6] | Campbell D (1899) Lectures on the Evolution of Plants. Kessinger Publishing, London. |
| [7] | Church AH (1919) Thalassiophyta and the Subaerial Transmigration. Oxford University Press, Oxford. |
| [8] | Clement M, Posada D, Crandall KA (2000) TCS: a computer program to estimate gene genealogies. Molecular Ecology, 9, 1657-1659. |
| [9] | Cranston KA, Hurwitz B, Ware D, Stein L, Wing RA (2009) Species trees from highly incongruent gene trees in rice. Systematic Biology, 58, 489-500. |
| [10] | Darriba D, Taboada GL, Doallo R, Posada D (2011) ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics, 27, 1164-1165. |
| [11] | Degnan JH, Rosenberg NA (2009) Gene tree discordance, phylogenetic inference and the multispecies coalescent. Trends in Ecology & Evolution, 24, 332-340. |
| [12] | Delwiche CF, Palmer JD (1996) Rampant horizontal transfer and duplication of Rubisco genes in eubacteria and plastids. Molecular Biology and Evolution, 13, 873-882. |
| [13] | Doolittle WF (1999) Phylogenetic classification and the universal tree. Science, 284, 2124-2128. |
| [14] | Dunn CW, Hejnol A, Matus DQ, Pang K, Browne WE, Smith SA, Seaver E, Rouse GW, Obst M, Edgecombe GD (2008) Broad phylogenomic sampling improves resolution of the animal tree of life. Nature, 452, 745-749. |
| [15] | Dunn CW, Howison M, Zapata F (2013) Agalma: an automated phylogenomics workflow. BMC Bioinformatics, 14, 330. |
| [16] | Eames AJ (1936) Morphology of Vascular Plants. McGraw- Hill Book Company, New York. |
| [17] | Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research, 32, 1792-1797. |
| [18] | Fritsch F (1945) Studies in the comparative morphology of the algae. IV. Algae and archegoniate plants. Annals of Botany, 9, 1-29. |
| [19] | Fritsch FE (1916) The algal ancestry of the higher plants. New Phytologist, 15, 233-249. |
| [20] | Fu L, Niu B, Zhu Z, Wu S, Li W (2012) CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics, 28, 3150-3152. |
| [21] | Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q (2011) Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nature Biotechnology, 29, 644-652. |
| [22] | Griffiths RC, Marjoram P (1996) Ancestral inference from samples of DNA sequences with recombination. Journal of Computational Biology, 3, 479-502. |
| [23] | Huson DH, Bryant D (2006) Application of phylogenetic networks in evolutionary studies. Molecular Biology and Evolution, 23, 254-267. |
| [24] | Huson DH, Scornavacca C (2011) A survey of combinatorial methods for phylogenetic networks. Genome Biology and Evolution, 3, 23-35. |
| [25] | Kenrick P, Crane PR (1997) The origin and early evolution of plants on land. Nature, 389, 33-39. |
| [26] | Li CS (1994) Origin of land plants is an important event of life evolution. Bulletin of National Natural Science Foundation of China, (4), 8-14. (in Chinese with English abstract) |
| [李承森 (1994) 生物进化的重大事件——陆地植物的起源及其研究的新进展. 中国科学基金, (4), 8-14.] | |
| [27] | Li L, Stoeckert CJ, Roos DS (2003) OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Research, 13, 2178-2189. |
| [28] | Mallet J (2005) Hybridization as an invasion of the genome. Trends in Ecology & Evolution, 20, 229-237. |
| [29] | Mirarab S, Bayzid MS, Warnow T (2016) Evaluating summary methods for multilocus species tree estimation in the presence of incomplete lineage sorting. Systematic Biology, 65, 366-380. |
| [30] | Mirarab S, Reaz R, Bayzid MS, Zimmermann T, Swenson MS, Warnow T (2014) ASTRAL: genome-scale coalescent- based species tree estimation. Bioinformatics, 30, 541-548. |
| [31] | Misof B, Liu S, Meusemann K, Peters RS, Donath A, Mayer C, Frandsen PB, Ware J, Flouri T, Beutel RG (2014) Phylogenomics resolves the timing and pattern of insect evolution. Science, 346, 763-767. |
| [32] | Nakhleh L (2010) Evolutionary phylogenetic networks: models and issues. In: Problem Solving Handbook in Computational Biology and Bioinformatics (eds Lenwood SH, Naren R), pp. 125-158. Springer, New York. |
| [33] | Nakhleh L (2013) Computational approaches to species phylogeny inference and gene tree reconciliation. Trends in Ecology & Evolution, 28, 719-728. |
| [34] | Regier JC, Shultz JW, Zwick A, Hussey A, Ball B, Wetzer R, Martin JW, Cunningham CW (2010) Arthropod relationships revealed by phylogenomic analysis of nuclear protein-coding sequences. Nature, 463, 1079-1083. |
| [35] | Rieseberg LH (1997) Hybrid origins of plant species. Annual Review of Ecology and Systematics, 28, 359-389. |
| [36] | Rubinstein CV, Gerrienne P, de la Puente G, Astini R, Steemans P (2010) Early Middle Ordovician evidence for land plants in Argentina (eastern Gondwana). New Phytologist, 188, 365-369. |
| [37] | Salichos L, Rokas A (2013) Inferring ancient divergences requires genes with strong phylogenetic signals. Nature, 497, 327-331. |
| [38] | Scott DH (1900) Studies in Fossil Botany. Adam & Charles Black, London. |
| [39] | Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM (2015) BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics, 31, 3210-3212. |
| [40] | Sneath PH (1975) Cladistic representation of reticulate evolution. Systematic Zoology, 24, 360-368. |
| [41] | Stamatakis A (2014) RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics, 30, 1312-1313. |
| [42] | Steemans P, Le Hérissé A, Melvin J, Miller MA, Paris F, Verniers J, Wellman CH (2009) Origin and radiation of the earliest vascular land plants. Science, 324, 353. |
| [43] | Sukumaran J, Holder MT (2010) DendroPy: a Python library for phylogenetic computing. Bioinformatics, 26, 1569-1571. |
| [44] | Syvanen M (1985) Cross-species gene transfer, implications for a new theory of evolution. Journal of Theoretical Biology, 112, 333-343. |
| [45] | Szöllősi GJ, Boussau B, Abby SS, Tannier E, Daubin V (2012) Phylogenetic modeling of lateral gene transfer reconstructs the pattern and relative timing of speciations. Proceedings of the National Academy of Sciences, USA, 109, 17513-17518. |
| [46] | Talavera G, Castresana J (2007) Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Systematic Biology, 56, 564-577. |
| [47] | Tofigh A, Hallett M, Lagergren J (2011) Simultaneous identification of duplications and lateral gene transfers. IEEE/ ACM Transactions on Computational Biology and Bioinformatics (TCBB), 8, 517-535. |
| [48] | Wellman CH, Osterloff PL, Mohiuddin U (2003) Fragments of the earliest land plants. Nature, 425, 282-285. |
| [49] | Wickett NJ, Mirarab S, Nguyen N, Warnow T, Carpenter E, Matasci N, Ayyampalayam S, Barker MS, Burleigh JG, Gitzendanner MA (2014) Phylotranscriptomic analysis of the origin and early diversification of land plants. Proceedings of the National Academy of Sciences, USA, 111, 4859-4868. |
| [50] | Woodhams MD, Lockhart PJ, Holland BR (2016) Simulating and summarizing sources of gene tree incongruence. Genome Biology and Evolution, 8, 1299-1315. |
| [51] | Zimmermann W (1938) Phylogenie. In: Manual of Pteridology (ed. Frans V), pp. 558-618. Springer, New York. |
| [52] | Zou XH, Ge S (2008) Conflicting gene trees and phylogenomics. Journal of Systematics and Evolution, 46, 795-807. (in Chinese with English abstract) |
| [邹新慧, 葛颂 (2008) 基因树冲突与系统发育基因组学研究. 植物分类学报, 46, 795-807.] |
| [1] | 朱瑞良, 马晓英, 曹畅, 曹子寅. 中国苔藓植物多样性研究进展[J]. 生物多样性, 2022, 30(7): 22378-. |
| [2] | 王伟, 刘阳. 植物生命之树重建的现状、问题和对策建议[J]. 生物多样性, 2020, 28(2): 176-188. |
| [3] | 杜新宇, 卢金梅, 李德铢. 石松类和蕨类植物质体基因组结构演化研究进展[J]. 生物多样性, 2019, 27(11): 1172-1183. |
| [4] | 常艳芬. 铁角蕨科的多倍化与物种多样性形成[J]. 生物多样性, 2017, 25(6): 621-626. |
| [5] | 曾丽萍, 张宁, 马红. 被子植物系统发育深层关系研究: 进展与挑战[J]. 生物多样性, 2014, 22(1): 21-39. |
| [6] | 鲁丽敏, 孙苗, 张景博, 李洪雷, 林立, 杨拓, 陈闽, 陈之端. 生命之树及其应用[J]. 生物多样性, 2014, 22(1): 3-20. |
| 阅读次数 | ||||||
|
全文 |
|
|||||
|
摘要 |
|
|||||
备案号:京ICP备16067583号-7
Copyright © 2022 版权所有 《生物多样性》编辑部
地址: 北京香山南辛村20号, 邮编:100093
电话: 010-62836137, 62836665 E-mail: biodiversity@ibcas.ac.cn