



草地占世界陆地面积的40%, 为人类提供了重要的生态系统服务, 如蓄水、固碳、减缓气候变化等(司绍诚等, 2022)。然而, 受气候变化及长期不合理的人为活动干扰, 全球范围内由于过度放牧导致超过23%的草地退化(Bardgett et al, 2021)。土壤微生物是草地生态系统健康的关键驱动因素, 通过调节养分循环、分解有机物、改良土壤结构、抑制植物病害和提高植物生产力, 发挥着一系列重要的生态作用(Coban et al, 2022; Liu et al, 2023)。研究证实草地退化可以通过改变土壤养分和植物特征来驱动土壤微生物多样性和功能的变化(Luo et al, 2020)。与未退化草地相比, 退化草地土壤细菌和真菌群落多样性、组成、生物网络和功能基因存在显著差异(Li et al, 2016; Luo et al, 2020, 2022)。土壤微生物群落在草地退化过程中可能具有双重作用(Xun et al, 2018; Coban et al, 2022)。一方面, 土壤微生物(如固氮菌、溶磷菌、丛枝菌根真菌等)可通过提高植物对环境胁迫的抗性来促进退化草地的恢复(Ezawa & Saito, 2018; Lai et al, 2021)。另一方面, 土壤传播的病原体会严重威胁植物的健康, 加剧草地退化(Che et al, 2019; Zhang Q et al, 2022)。然而, 这些研究主要集中在细菌和真菌, 对原生生物群落的研究很少, 限制了我们对退化草地生态系统土壤微生物群落生态学的整体认识。
原生生物作为土壤微生物的重要组成部分, 种类丰富, 功能多样(Singer et al, 2021)。原生生物是细菌、真菌和其他小型真核生物的主要捕食者, 在微生物食物网中扮演着关键角色(Xiong et al, 2018)。原生生物的捕食活动促进了固定在微生物生物量中营养物质的释放, 促进了营养物质的周转和吸收, 影响了生态系统的功能。另外, 原生生物还可以通过调节有机质降解和碳固定来促进养分循环(Jassey et al, 2015; Kramer et al, 2016; Geisen et al, 2018)。最近的研究报道了原生生物在植物疾病控制和促进植物生长中的作用(Xiong et al, 2020), 可能是通过直接捕食土壤传播的病原体(Geisen et al, 2018), 或间接改变植物有益微生物和次生代谢物的产生(Gao et al, 2019)。长期过度放牧显著影响了青藏高原高寒草甸土壤原生生物功能群的组成及其生物或非生物调节过程(Lin et al, 2017)。亚高山草甸土壤原生生物群落组成和结构在海拔梯度上存在显著的差异, 土壤含水量、总氮和植物丰富度指数是主要的环境驱动因子(罗正明等, 2021)。作为指示生物, 原生生物同其他微生物一样在草地生态系统中起着关键作用, 且对环境变化非常敏感, 能较早地指示土壤生态系统功能的变化(Zhao et al, 2019)。因此, 阐明草地退化过程中原生生物群落多样性、结构和功能变化及其驱动机制至关重要。近年来, 随着分子技术的快速发展和原生生物数据库的不断完善, 基于18S rDNA高通量测序技术方法成为全面分析原生生物群落组成和多样性的重要手段(Geisen et al, 2018)。
五台山被称为“华北屋脊”, 位于太行山北端, 最高峰叶斗峰海拔3,061.1 m, 植被垂直带谱明显, 海拔从高到低依次为: 亚高山和高山草甸带-亚高山灌丛带-针叶林带-针、阔叶混交林带-山地灌丛带-灌草丛及农田带。分布在五台山海拔2,000-3,000 m间的亚高山草甸是华北地区面积最大(106,993 ha)的高寒草甸之一(罗正明等, 2022)。五台山亚高山草甸群落以耐寒冷、密丛短根茎地下芽嵩草(Kobresia)以及苔草(Carex)、禾草为建群种。土壤类型为亚高山草甸土, 又名黑毡土, 是在高寒(年均气温−4℃)、多水(年降雨量900 mm以上)、长日照(年均2,722.8 h)、大风、矮化草甸植被的作用下, 经反复冻融发育而成, 草甸地表呈“草丛土丘”景观。五台山亚高山草甸是华北地区重要的生态屏障和夏季牧场。然而, 随着人类活动加剧, 加上连年超载放牧, 使得五台山亚高山草甸植被遭到严重破坏, 生物多样性锐减, 面临严重的退化问题。据统计不同退化程度的草甸约占五台山草甸总面积的3/5, 中度以上退化的草甸占整体退化草甸的一半以上(Luo et al, 2020)。本研究以五台山典型亚高山草甸为研究对象, 通过18S rDNA高通量测序, 对4个不同退化阶段(未退化(nondegraded, ND)、轻度(lightly degraded, LD)、中度(moderately degraded, MD)和重度退化(heavily degraded, HD))亚高山草甸土壤原生生物群落进行了分析。旨在探讨: (1)不同退化阶段亚高山草甸土壤原生生物群落结构组成和多样性变化特征; (2)土壤原生生物群落结构变化的主要驱动因子。研究结果不仅可以提供亚高山草甸土壤原生生物多样性方面的基础资料, 深入理解原生生物对于土壤微生态系统中的影响和作用, 还能为亚高山草甸生态系统监测与保护提供科学依据。
1 材料与方法
1.1 样地设置与样品采集
图1
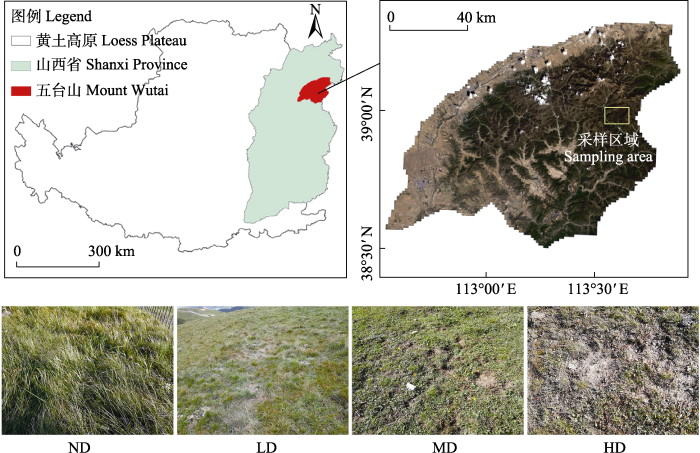
图1
五台山采样位置和区域示意图。ND: 未退化草甸; LD: 轻度退化草甸; MD: 中度退化草甸; HD: 重度退化草甸。
Fig. 1
Sketch map of sampling location and area of Mount Wutai. ND, Nondegraded meadow; LD, Lightly degraded meadow; MD, Moderately degraded meadow; HD, Heavily degraded meadow.
1.2 环境因子测定
记录样方内植物种类、物种数、群落总盖度和高度。植物多样性测定采用Shannon-Wiener指数和丰富度指数进行评价。采用烘干法测定土壤含水量, 电位法(HANNA, 意大利)测定土壤pH值(土水比为1∶2.5), 元素分析仪(Elementar Vario MACRO, 德国)测定总碳和总氮, K2Cr2O7氧化法测定土壤有机碳, 间断元素分析仪(Clever Chem 380, 德国)测定铵态氮、硝态氮、亚硝态氮, 钼蓝法测定土壤有效钾, 火焰光谱法测定土壤有效磷, 激光粒度仪法测定土壤粒径。
1.3 高通量测序及生物信息学分析
使用土壤DNA试剂盒(Omega Bio-tek, USA)从0.25 g土壤中提取总DNA。用正向引物TAReuk 454FWD1F (5°-CCAGCASCYGCGGTAATTCC-3°) 和反向引物TAReukREV3F (5°-ACTTTCGTTCTT GATYRA-3°)扩增18S rRNA的V4高变区(Stoeck et al, 2010)。PCR扩增程序: 94℃初始变性3 min, 然后在95℃变性30 s, 55℃退火30 s, 72℃延伸45 s, 共进行27个循环, 最后在72℃延伸10 min。PCR产物在Illumina Miseq平台(上海美吉生物医药科技有限公司)上进行纯化、聚合和测序。利用QIIME2中的标准操作程序对原始序列进行分析。使用UPARSE进行嵌合体去除, 按97%序列相似度聚类OTUs。根据Protist Ribosomal Reference (PR2)数据库对基于真核生物18S rRNA基因数据的原生生物序列进行分类(Guillou et al, 2013)。去除被定义为真菌、后生动物(Metazoan)、红藻(Rhodophyta)和链形植物(Streptophyta)的OTUs (占所有OTUs的43.5%), 按最小样本序列数抽平, 然后进行后续的分析(Zhao et al, 2019; Zhang SY et al, 2022)。平均相对丰度 > 1%,的门被定义为优势门。
1.4 数据分析
利用QIIME2软件计算原生生物群落的α多样性。采用线性判别分析(linear discriminant analysis, LDA)结合LEfSe分析确定不同退化阶段亚高山草甸间统计学差异的生物标志物(biomarker) (P < 0.05和LDA评分 > 3.5)。使用基于Bray-Curtis距离的非度量多维尺度分析(non-metric multidimensional scaling analysis, NMDS)评估原生生物群落的整体结构变化。使用相似性分析(analysis of similarities, ANOSIM)来确定土壤原生生物群落结构差异的显著性。采用Pearson相关分析和Mantel test分别分析环境因子间相关性及环境与土壤原生生物群落结构的关系。进一步使用逐步回归和Monte Carlo permutation test对所有环境变量进行前选择, 选择具有统计学意义(P < 0.05)的环境变量进行冗余分析(redundancy analysis, RDA), 评价原生生物群落结构与环境变量之间的相关性。基于前选择筛选的环境因子, 采用方差分解分析(variance partitioning analysis, VPA)定量土壤理化性质和植物变量对原生生物群落结构变化的影响程度。采用单因素方差分析和Duncan多重比较分析对不同退化阶段亚高山草甸土壤原生生物优势门相对丰度以及原生生物群落α多样性和β多样性相异性进行显著性差异检验。基于Galaxy进行LEfSe的在线分析及绘图(
2 结果
2.1 不同退化阶段亚高山草甸土壤原生生物群落组成的变化
扩增子测序结果显示, 4个不同退化阶段亚高山草甸土壤的所有样本中共鉴定出4,726个OTUs。有孔虫界(Rhizaria)是在所有样本中相对丰度最高的超群(占所有序列数的49.52%), 其他超群按相对丰度从高到低依次为不等鞭毛类(Stramepopila, 19.29%)、囊泡虫类(Alveolata, 17.14%)、变形虫界(Amoebozoa, 10.12%)、泛植物界(Archaeplastida, 2.17%)、后鞭毛生物(Opisthokonta, 0.64%)、古虫界(Excavata, 0.58%)、Hacrobia (0.32%)和无根虫界(Apusozoa, 0.21%)。一共包含25门61纲104目176科298属。其中丝足门(Cercozoa)、褐藻门(Ochrophyta)、纤毛门(Ciliophora)、叶足亚门(Lobosa)、不等鞭毛类中未定义的门(Stramepopila_X)、锥足亚门(Conosa)、绿藻门(Chlorophyta)和顶复门(Apicomplexa)为优势门(图2a)。纤毛门、叶足亚门、绿藻门、Choanoflagellida和Perkinsea在4个不同退化阶段亚高山草甸间存在显著差异(P < 0.05, 图2b)。
图2
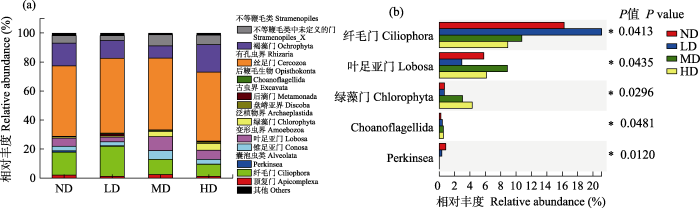
图2
不同退化阶段亚高山草甸土壤原生生物优势门(a)和存在显著差异优势门(b)的相对丰度。ND: 未退化草甸; LD: 轻度退化草甸; MD: 中度退化草甸; HD: 重度退化草甸。P值表示不同退化阶段亚高山草甸间土壤原生生物门相对丰度的差异水平, *表示存在显著差异(P < 0.05)。
Fig. 2
Relative abundance of dominant (a) and significantly differentially dominant (b) phyla of soil protist in subalpine meadow at different degradation stages. ND, Nondegraded meadow; LD, Lightly degraded meadow; MD, Moderately degraded meadow; HD, Heavily degraded meadow. P value indicates the level of difference in relative abundance of soil protists among subalpine meadows at different stages of degradation, and * indicates a significant difference (P < 0.05).
LEfSe结果显示, 不同退化阶段亚高山草甸共有32个相对丰度显著较高的原生生物分类单元(LDA > 3.5, P < 0.05, 图3)。ND草甸中富集了9个原生生物类群, 即在超群水平的类群为囊泡虫类(Alveolata); 在门水平上有Perkinsea; 纲水平上有Perkinsida和Endomyxa-Phytomyxea; 目水平上有Perkinsida中未定义的目(Perkinsida_X)和质体目(Plasmodiophorida); 在科水平上有Perkinsida中未定义的科(Perkinsida_XX)、Polymyxa_linege和Paracercomonadidae。LD草甸中富集了盾纤目(Scuticociliatida)、精巢虫科(Orchitophryidae)和Allapsidae科。MD草甸中检测到10个显著差异的原生生物类群(2门2纲3目3科), 即叶足亚门(Lobosa)、变形虫纲(Tubulinea)、Echinamoebida (目)、Vermamoebidae (目)、ATCC50593-lineage (科)、ATCC50593-Flamella (科)、不等鞭毛类中未定义的门(Stramenopiles_X)、卵菌纲(Oomycota)、卵菌纲中未定义的目(Oomycota_X)和Peronosporales (科)。HD草甸富集了10个原生生物分类群(2超群1门3纲2目2科), 包括泛植物界超群(Archaeplastida)、绿藻门(Chlorophyta)、绿藻纲(Chlorophyceae)、共球藻纲(Trebouxiophyceae)、环藻目(Sphaeropleales)、环藻目中未定义的科(Sphaeropleales_X)、不等鞭毛类超群(Stramenopiles)、硅藻纲(Bacillariophyta)、硅藻纲中未定义的目(Bacillariophyta_X)和Raphid-pennate (科)。结果表明, 亚高山草甸退化过程中土壤原生生物群落组成和相对丰度在各分类水平上发生了显著变化。
图3
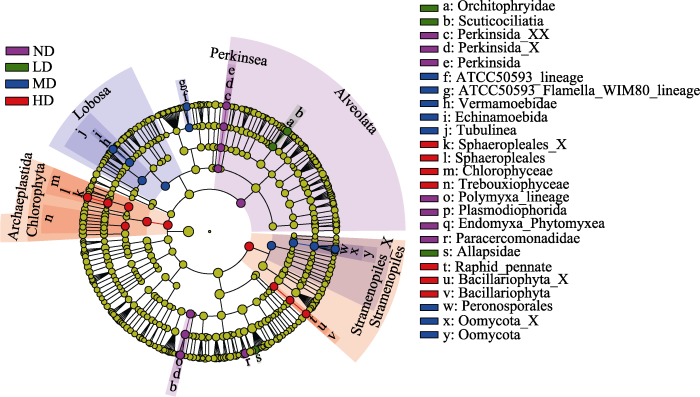
图3
不同退化阶段亚高山草甸土壤原生生物群落差异的LEfSe分析。ND: 未退化草甸; LD: 轻度退化草甸; MD: 中度退化草甸; HD: 重度退化草甸。每个圆环为一个分类学层次内的所有分类群, 从内到外的圆环分别代表超群、门、纲、目和科; 圆环上的节点表示分类学层次上的一个分类单元, 每个节点的直径与丰度成正比; 不同退化程度草甸中相对丰度显著较高的分类单元(生物标志物)在进化分支图中进行了颜色编码。
Fig. 3
LEfSe analysis showing soil protist community differences in subalpine meadow at different degradation stages. ND, Nondegraded meadow; LD, Lightly degraded meadow; MD, Moderately degraded meadow; HD, Heavily degraded meadow. Each circular ring deposit all taxa within a taxonomic level, the circular ring from inside to outside represents supergroup, phylum, class, order and family, respectively. The node on the circular ring represents taxon, affiliating within the taxonomic level. The diameter of each node is proportional to the abundance of the group. Taxa that had significantly higher relative abundance in a certain treatment within each meadow degradation type were color-coded within the cladogram.
2.2 不同退化阶段亚高山草甸土壤原生生物群落多样性的变化
图4
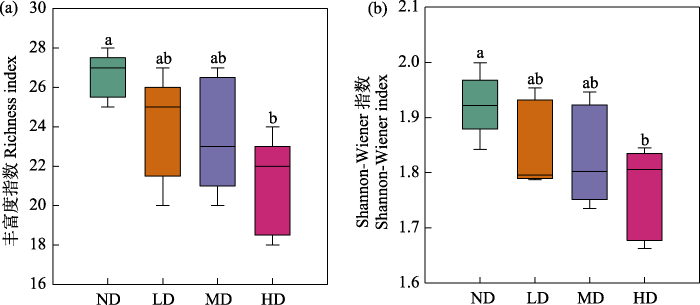
图4
不同退化阶段亚高山草甸土壤原生生物群落α多样性。ND: 未退化草甸; LD: 轻度退化草甸; MD: 中度退化草甸; HD: 重度退化草甸。各组间不相同的小写字母表示存在显著差异(P < 0.05)。
Fig. 4
The α diversity indices of soil protist communities in subalpine meadow at different degradation stages. ND, Nondegraded meadow; LD, Lightly degraded meadow; MD, Moderately degraded meadow; HD, Heavily degraded meadow. Data that do not share a lowercase letter are significantly different (P < 0.05).
图5
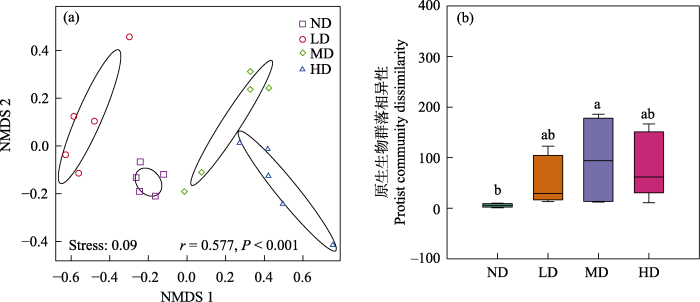
图5
不同退化阶段亚高山草甸土壤原生生物群落非度量多维尺度分析(NMDS) (a)及相异性分析(基于Bray-Curtis距离) (b)。r = 0.577, P < 0.001为不同退化阶段亚高山草甸之间原生生物群落相似性的ANOSIM检验结果。ND: 未退化草甸; LD: 轻度退化草甸; MD: 中度退化草甸; HD: 重度退化草甸。
Fig. 5
Non-metric multidimensional scaling (NMDS) (a) and dissimilarity analysis (based on Bray-Curtis distance) (b) of soil protist communities in subalpine meadow at different degradation stages. The r = 0.577, P < 0.001 are the ANOSIM test results of protist community similarity between subalpine meadows in different degradation stages. ND, Nondegraded meadow; LD, Lightly degraded meadow; MD, Moderately degraded meadow; HD, Heavily degraded meadow.
2.3 环境因子对土壤原生生物群落结构的影响
Mantel test分析表明, 土壤含水量、全氮、总碳、硝态氮、植物盖度会显著影响原生生物群落结构(图6, P < 0.01)。绿藻门与土壤质地(粘粒、粉粒和砂粒)、pH、碳氮比、植物盖度和高度显著相关(P < 0.05); 叶足亚门与土壤粉粒、pH、植物盖度和丰富度显著相关(P < 0.05); 纤毛门与pH和植物盖度显著相关(图6, P < 0.05)。RDA结果显示, 轴1和轴2分别解释了原生生物群落结构变异量的31.87%和22.30%, 土壤和植物参数共同解释了原生生物群落变异量的54.17% (图7a)。总氮、植物Shannon-Wiener指数、地上生物量、土壤含水量和铵态氮是驱动原生生物群落结构变化的重要环境因子(P < 0.05)。方差分解分析结果显示, 土壤理化因子(总氮、土壤含水量和铵态氮)和植被参数(植物Shannon-Wiener指数和地上生物量)共同解释了原生生物群落变异的38.44%, 其中土壤理化因子单独解释了20.69%, 植被参数单独解释了7.85%, 土壤理化因子对群落结构变异的贡献大于植被参数(图7b)。
图6
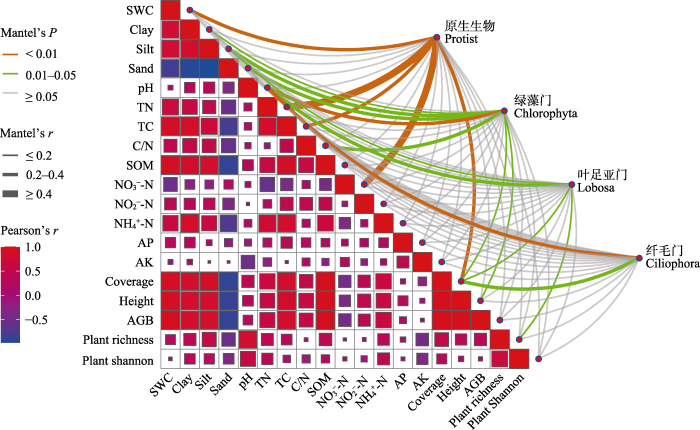
图6
环境因子相关性及环境与土壤原生生物群落关系的Mantel分析。SWC: 土壤含水量; Clay: 粘粒含量; Silt: 粉粒含量; Sand: 砂粒; TN: 全氮; TC: 总碳; C/N: 碳氮比; SOM: 土壤有机质; NO3--N: 硝态氮; NO2--N: 亚硝态氮; NH4+-N: 铵态氮; AP: 有效磷; AK: 有效钾; Coverage: 植被盖度; Height: 植被高度; AGB: 地上生物量; Plant richness: 植物丰富度指数; Plant Shannon: 植物Shannon-Wiener指数。
Fig. 6
Correlation of environmental factors and Mantel analysis of relationship between environment and soil protist communities. SWC, Soil water content; Clay, Clay content; Silt, Silt content; Sand, Sand content; TN, Total nitrogen; TC, Total carbon; C/N, Carbon nitrogen ratio; SOM, Soil organic matter; NO3--N, Nitrate nitrogen; NO2--N, Nitrous nitrogen; NH4+-N, Ammonium nitrogen; AP, Available phosphorus; AK, Available potassium; Coverage, Vegetation coverage; Height, Vegetation height; AGB, Aboveground biomass; Plant richness: Plant richness index; Plant Shannon, Plant Shannon-Wiener index.
图7
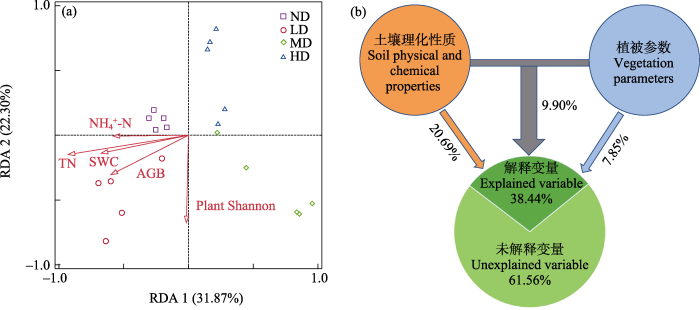
图7
土壤原生生物群落结构与环境变量的冗余分析(a)和方差分解分析(b)。TN: 总氮; Plant Shannon: 植物Shannon- Wiener指数; AGB: 地上生物量; SWC: 土壤含水量; NH4+-N: 铵态氮。ND: 未退化草甸; LD: 轻度退化草甸; MD: 中度退化草甸; HD: 重度退化草甸。
Fig. 7
Relationships between soil protist community structure and environmental variables based on redundancy analysis (a) and variation partitioning analysis (b). TN, Total nitrogen; Plant Shannon, Plant Shannon-Wiener index; AGB, Aboveground biomass; SWC, Soil water content; NH4+-N, Ammonium nitrogen. ND, Nondegraded meadow; LD, Lightly degraded meadow; MD, Moderately degraded meadow; HD, Heavily degraded meadow.
3 讨论
3.1 土壤原生生物群落物种组成对亚高山草甸退化的响应
通过原生生物群落门水平的组成和差异分析结果可知, 不同退化阶段亚高山草甸中土壤原生生物群落的优势类群基本一致(图2), 其中丝足门、褐藻门、纤毛门相对丰度最高, 与黑土中的优势门相似, 而红壤中以绿藻门和丝足门为主(Zhao et al, 2019)。这可能是由于亚高山草甸土和黑土中有机质含量通常比红壤高, 成土气候条件也明显不同。纤毛门、绿藻门、Choanoflagellida和Perkinsea的相对丰度在不同退化阶段亚高山草甸中存在显著差异(图3, P < 0.05)。另外, 4种不同退化阶段亚高山草甸均富集了不同的生物标志物(图3), 说明土壤原生生物群落组成和相对丰度沿草甸退化梯度呈现出明显的分异。未退化草甸富集的Perkinsea被认为是囊泡虫超群中唯一的寄生群, 广泛分布在从海洋到淡水的各种水生环境和湿地中(Itoïz et al, 2022)。Perkinsea由寄生原生生物的4个主要谱系组成, 寄主范围广泛, 其中一些因较强的地理入侵能力和致病性而构成重大生态和经济威胁。与未退化草甸相比, 退化草甸中Perkinsea的相对丰度显著减少, 其原因可能是未退化草甸中含水量高, 更适合喜湿的Perkinsea生存(Chambouvet et al, 2014)。Perkinsea可以寄生在各种寄主(动物、植物或其他生物)上, 未退化草甸土壤养分含量高以及植被状态良好, 为它们提供了最丰富的寄主来源, 这可能是它们更多的存在于未退化草甸的另一个原因(Sun et al, 2021)。然而, 目前对Perkinsea的形态、生命周期、宿主的身份和生理特征仍然认识不够, 特别是它们土壤中的相关信息知之甚少(Jeon & Park, 2021)。轻度草甸中富集的盾纤目(Scuticociliatida)纤毛虫通常个体较小, 大多数在15-50 μm范围内, 广泛生存于各类水环境中, 尤其在富营养化水体中是常见的自由生活(或兼性寄生)原生动物。中度退化草甸中富集的叶足亚门属于变形虫界超群, 变形虫门中多数物种靠细胞内原生质的流动而移动(Zhang SY et al, 2022)。其伪足类似于手指形状, 边缘是钝的, 生活于水、泥土或腐败有机物中, 滋养体以细菌为主, 二分裂方式增殖, 并可形成包囊, 有些也被发现与其他生物共生, 某些甚至是病原体(Sun et al, 2021)。另外, 中度退化草甸中富集的卵菌纲属于不等鞭毛类, 具有渗透性和溶氧性, 可以自由生活, 兼性或专性寄生于其他卵菌、真菌、植物和动物, 并通过溶氧作用促进有机物的分解(Geisen et al, 2018)。卵菌中部分成员属于植物致病菌, 可能会对中度退化草甸植物产生不利的影响。重度退化草甸富集了包含绿藻门的泛植物界超群和包含硅藻纲的不等鞭毛类超群, 说明重度退化草甸富集的原生生物主要是光合自养类群。与未退化草甸相比, 中度和重度退化草甸中光合原生生物绿藻门的丰度显著增加(图2)。草地退化降低了净初级生产力, 进而导致亚高山草甸中土壤肥力和有机碳含量显著下降。然而, 退化严重草甸中相对丰度较高的光合自养原生生物可以通过光合作用获取能量输入到土壤(Jassey et al, 2015)。因此, 光合自养原生生物可以作为初级生产者对退化严重草甸中土壤有机碳固存做出贡献, 在一定程度上可以缓解退化对草地生态系统的不利影响。然而, 在退化亚高山草甸生态系统中地上植物和光合自养类群原生生物对初级生产力的相对贡献还需要进一步深入研究。
3.2 土壤原生生物群落多样性对亚高山草甸退化的响应
亚高山草甸土壤原生生物群落的α多样性随退化加剧呈下降的趋势, 特别是重度退化草甸土壤原生生物群落α多样性显著下降(图4)。这与前期发现的土壤真菌群落α多样性变化趋势相一致(罗正明等, 2022), 土壤细菌群落α多样性没有发生显著的变化(Luo et al, 2020)。这说明土壤微生物组中不同组分(如细菌、真菌和原生生物)的α多样性对亚高山草甸退化的响应模式不同, 原生生物群落和真菌群落α多样性比细菌群落对草甸退化更敏感。原生生物和真菌属于真核生物, 细菌属于原核生物, 由于两者结构和生理的差异, 导致真核生物在草地退化过程中对土壤养分限制和植被退化的敏感性高于原核生物(Lauber et al, 2008), 这可能是导致这一观察结果的原因。另一方面, 原生生物的栖息地生态位宽度低于细菌(Wu et al, 2018), 这表明原生生物对环境变化的耐受性低于细菌。草地退化导致原生生物多样性的丧失可能随后对土壤生态系统稳定性和土壤多功能产生负面影响(Wu et al, 2022)。
亚高山草甸退化显著影响了土壤原生生物群落结构和增加了β多样性相异性(图5), 与前期研究细菌和真菌的结果一致(Luo et al, 2020; 罗正明等, 2022), 说明土壤微生物群落结构对亚高山草甸退化较为敏感。原生生物群落结构沿草甸退化梯度的变化(r = 0.577, P < 0.001)比细菌(r = 0.421, P < 0.001)和真菌群落结构(r = 0.445, P < 0.001)的变化更为明显(Luo et al, 2020; 罗正明等, 2022)。4个不同退化阶段亚高山草甸中任意两个土壤原生生物群落组成均显著分离(P < 0.05), 而细菌和真菌群落组成在中度和重度退化草甸间没有显著差异。这些结果表明原生生物群落结构的变化比细菌和真菌更为敏感。特别是本研究发现纤毛门对草地退化较敏感, 与未退化和轻度退化草甸相比, 中度和重度退化草甸中纤毛门的相对丰度显著下降。宁应之等(2018)研究发现退耕还林生态恢复后, 土壤纤毛虫的优势类群从肾形目逐渐演替为散毛目, 从r-对策型演替成K-对策型, 土壤纤毛虫群落结构可作为对退耕还林生态恢复的评价指标。土壤纤毛虫是原生生物中的重要类群之一, 其体积小、物种丰富、生长周期短、分布广泛、群落演替迅速, 对环境变化敏感(Zhao et al, 2019), 本研究结果证实了纤毛虫原生生物群落作为指示环境变化指标的潜力, 在未来的工作中应该优先考虑, 例如, 构建纤毛虫相对丰度与草地土壤退化的关系曲线, 以指示草地土壤退化程度等。
3.3 亚高山草甸退化过程中土壤原生生物群落结构变化与环境变量的关系
亚高山草甸退化降低了土壤中的养分、总碳、总氮和有机质含量, 这是由于退化导致植物生物量和土壤团聚体减少, 可能会增加养分淋滤, 导致养分的损失(Luo et al, 2022)。草甸退化过程中植物和土壤等环境变量发生了明显的变化, 不可避免地影响着土壤原生生物群落结构。土壤理化因子对原生生物群落组成变化的解释度(20.69%)高于植物参数(7.85%), 说明土壤理化性质可能是决定亚高山草甸退化土壤原生生物群落变化的关键因素。本研究发现总氮、植物Shannon-Wiener指数、地上生物量、土壤含水量和铵态氮显著影响了土壤原生生物群落结构(P < 0.05), 是最重要的环境驱动因子。与之前的报道一致, 土壤中氮和其他营养物质塑造了土壤原生生物多样性和结构。纤毛虫、有壳变形虫(testate amoebae)和藻类(algae)的组成、多样性和密度随土壤氮梯度变化很大(Clarholm, 2002; Acosta-Mercado & Lynn, 2004; Krashevska et al, 2014)。Hu等(2022)在青藏高寒草甸中发现提升30%降水量增加了吞噬性原生生物的多样性, 减少30%降水量降低了吞噬性原生生物的多样性, 且氮输入(12 g N·m−2·yr−1)可以增加原生生物的丰度, 通过增加微生物生物量抵消降水减少对吞噬型原生生物相对丰度的负面影响。水分对土壤原生生物非常重要, 原生生物基本上依赖于连接土壤孔隙的水层来移动、进食和繁殖, 它们的栖息地大小将随着土壤水分有效性的变化而增大或缩小(Geisen et al, 2014)。原生生物数量最多的时候往往出现在潮湿的季节和雨后。因此, 亚高山草甸退化过程中土壤水分含量显著减少, 可能会影响土壤原生生物群落组成和结构。植物和植被类型可以通过各种方式影响土壤原生生物群落(Geisen et al, 2018), 包括凋落物或根系分泌物质量的差异、小气候的变化(如开阔地与林地)以及对细菌或真菌群落的影响。植物与其微生物群落密切相关, 尤其是在根际。Acosta-Mercado和Lynn (2006)的研究发现两种不同热带植物根际差异显著影响了其纤毛虫群落的结构和组成。较高的植物功能多样性被证实可以增加变形虫原生生物的丰度。沿冰川消退带有壳变形虫的物种丰富度沿时间序列增加, 与植物群落演替一致(Carlson et al, 2010)。然而, 变形虫物种丰富度的增加并不一定与维管植物丰富度相关, 因为其他因素如土壤有机碳或氮含量也有助于增加原生生物多样性(Dassen et al, 2017)。亚高山草甸退化过程中原生生物群落对生物和非生物驱动因素的复杂组合做出反应, 这些驱动因素的相互作用仍远未被理解, 有待进一步深入研究。
4 结论
本研究利用高通量测序技术对五台山不同退化阶段亚高山草甸土壤原生生物群落多样性和结构特征进行研究。研究结果表明, 亚高山草甸退化显著改变了土壤原生生物群落组成和部分类群的相对丰度。在门水平上, 纤毛门、绿藻门、Choanoflagellida和Perkinsea对草甸退化最敏感。不同退化阶段亚高山草甸富集了不同的生物标志物, 且土壤原生生物群落结构存在显著差异。随着亚高山草甸退化加剧, 土壤原生生物群落α多样性呈下降的趋势。与未退化草甸相比, 重度退化草甸土壤原生生物群落α多样性显著下降。土壤原生生物群落对亚高山草甸退化过程中土壤环境条件的变化有较好的响应。总氮、植物Shannon-Wiener指数、地上生物量、土壤含水量和铵态氮是原生生物群落结构变化的主要驱动因素。土壤理化性质和植物参数均对土壤原生生物群落结构的变化产生了重大影响, 且土壤理化性质对原生生物群落结构的影响大于植被参数的影响。
参考文献
Soil ciliate species richness and abundance associated with the rhizosphere of different subtropical plant species
DOI:10.1111/jeu.2004.51.issue-5 URL [本文引用: 1]
Contrasting soil ciliate species richness and abundance between two tropical plant species: A test of the plant effect
DOI:10.1007/s00248-006-9067-3
PMID:16645921
[本文引用: 1]

We still have a rudimentary understanding about the mechanism by which plant roots may stimulate soil microbial interactions. A biochemical model involving plant-derived biochemical fractions, such as exudates, has been used to explain this "rhizosphere effect" on bacteria. However, the variable response of other soil microbial groups, such as protozoa, to the rhizosphere suggests that other factors could be involved in shaping their communities. Thus, two experiments were designed to: (1) determine whether stimulatory and/or inhibiting factors associated with particular plant species regulate ciliate diversity and abundance and (2) obtain a better understanding about the mechanism by which these plant factors operate in the rhizosphere. Bacterial and chemical slurries were reciprocally exchanged between two plant species known to differ in terms of ciliate species richness and abundance (i.e., Canella winterana and plantation Tectona grandis). Analysis of variance showed that the bacteria plus nutrients and the nutrients only treatment had no significant effect on overall ciliate species richness and abundance when compared to the control treatment. However, the use of only colpodean species increased the taxonomic resolution of treatment effects revealing that bacterial slurries had a significant effect on colpodean ciliate species richness. Thus, for particular rhizosphere ciliates, biological properties, such as bacterial diversity or abundance, may have a strong influence on their diversity and possibly abundance. These results are consistent with a model of soil bacteria-mediated mutualisms between plants and protozoa.
Combatting global grassland degradation
Community development along a proglacial chronosequence: Are above-ground and below-ground community structure controlled more by biotic than abiotic factors?
DOI:10.1111/jec.2010.98.issue-5 URL [本文引用: 1]
Diverse molecular signatures for ribosomally ‘active’ Perkinsea in marine sediments
DOI:10.1186/1471-2180-14-110
PMID:24779375
[本文引用: 1]
Background: Perkinsea are a parasitic lineage within the eukaryotic superphylum Alveolata. Recent studies making use of environmental small sub-unit ribosomal RNA gene (SSU rDNA) sequencing methodologies have detected a significant diversity and abundance of Perkinsea-like phylotypes in freshwater environments. In contrast only a few Perkinsea environmental sequences have been retrieved from marine samples and only two groups of Perkinsea have been cultured and morphologically described and these are parasites of marine molluscs or marine protists. These two marine groups form separate and distantly related phylogenetic clusters, composed of closely related lineages on SSU rDNA trees. Here, we test the hypothesis that Perkinsea are a hitherto under-sampled group in marine environments. Using 454 diversity 'tag' sequencing we investigate the diversity and distribution of these protists in marine sediments and water column samples taken from the Deep Chlorophyll Maximum (DCM) and sub-surface using both DNA and RNA as the source template and sampling four European offshore locations. Results: We detected the presence of 265 sequences branching with known Perkinsea, the majority of them recovered from marine sediments. Moreover, 27% of these sequences were sampled from RNA derived cDNA libraries. Phylogenetic analyses classify a large proportion of these sequences into 38 cluster groups (including 30 novel marine cluster groups), which share less than 97% sequence similarity suggesting this diversity encompasses a range of biologically and ecologically distinct organisms. Conclusions: These results demonstrate that the Perkinsea lineage is considerably more diverse than previously detected in marine environments. This wide diversity of Perkinsea-like protists is largely retrieved in marine sediment with a significant proportion detected in RNA derived libraries suggesting this diversity represents ribosomally 'active' and intact cells. Given the phylogenetic range of hosts infected by known Perkinsea parasites, these data suggest that Perkinsea either play a significant but hitherto unrecognized role as parasites in marine sediments and/or members of this group are present in the marine sediment possibly as part of the ` seed bank' microbial community.
Degraded patch formation significantly changed microbial community composition in alpine meadow soils
DOI:10.1016/j.still.2019.104426 URL [本文引用: 1]
Bacteria and protozoa as integral components of the forest ecosystem—Their role in creating a naturally varied soil fertility
DOI:10.1023/A:1020543424098 URL [本文引用: 1]
Soil microbiota as game-changers in restoration of degraded lands
Differential responses of soil bacteria, fungi, Archaea and protists to plant species richness and plant functional group identity
How do arbuscular mycorrhizal fungi handle phosphate? New insight into fine-tuning of phosphate metabolism
DOI:10.1111/nph.15187
PMID:29701874
[本文引用: 1]
Contents Summary 1116 I. Introduction 1116 II. Foraging for phosphate 1117 III. Fine-tuning of phosphate homeostasis 1117 IV. The frontiers: phosphate translocation and export 1119 V. Conclusions and outlook 1120 Acknowledgements 1120 References 1120 SUMMARY: Arbuscular mycorrhizal fungi form symbiotic associations with most land plants and deliver mineral nutrients, in particular phosphate, to the host. Therefore, understanding the mechanisms of phosphate acquisition and delivery in the fungi is critical for full appreciation of the mutualism in this association. Here, we provide updates on physical, chemical, and biological strategies of the fungi for phosphate acquisition, including interactions with phosphate-solubilizing bacteria, and those on the regulatory mechanisms of phosphate homeostasis based on resurveys of published genome sequences and a transcriptome with reference to the latest findings in a model fungus. For the mechanisms underlying phosphate translocation and export to the host, which are major research frontiers in this field, not only recent advances but also testable hypotheses are proposed. Lastly, we briefly discuss applicability of the latest tools to gene silencing in the fungi, which will be breakthrough techniques for comprehensive understanding of the molecular basis of fungal phosphate metabolism.© 2018 The Authors. New Phytologist © 2018 New Phytologist Trust.
Protists: Puppet masters of the rhizosphere microbiome
DOI:S1360-1385(18)30244-9
PMID:30446306
[本文引用: 1]
The rhizosphere microbiome is a central determinant of plant performance. Microbiome assembly has traditionally been investigated from a bottom-up perspective, assessing how resources such as root exudates drive microbiome assembly. However, the importance of predation as a driver of microbiome structure has to date largely remained overlooked. Here we review the importance of protists, a paraphyletic group of unicellular eukaryotes, as a key regulator of microbiome assembly. Protists can promote plant-beneficial functions within the microbiome, accelerate nutrient cycling, and remove pathogens. We conclude that protists form an essential component of the rhizosphere microbiome and that accounting for predator-prey interactions would greatly improve our ability to predict and manage microbiome function at the service of plant growth and health.Copyright © 2018 Elsevier Ltd. All rights reserved.
Soil water availability strongly alters the community composition of soil protists
DOI:10.1016/j.pedobi.2014.10.001 URL [本文引用: 1]
Methodological advances to study the diversity of soil protists and their functioning in soil food webs
DOI:10.1016/j.apsoil.2017.05.021 URL [本文引用: 5]
Soil protists: A fertile frontier in soil biology research
DOI:10.1093/femsre/fuy006
PMID:29447350
Protists include all eukaryotes except plants, fungi and animals. They are an essential, yet often forgotten, component of the soil microbiome. Method developments have now furthered our understanding of the real taxonomic and functional diversity of soil protists. They occupy key roles in microbial foodwebs as consumers of bacteria, fungi and other small eukaryotes. As parasites of plants, animals and even of larger protists, they regulate populations and shape communities. Pathogenic forms play a major role in public health issues as human parasites, or act as agricultural pests. Predatory soil protists release nutrients enhancing plant growth. Soil protists are of key importance for our understanding of eukaryotic evolution and microbial biogeography. Soil protists are also useful in applied research as bioindicators of soil quality, as models in ecotoxicology and as potential biofertilizers and biocontrol agents. In this review, we provide an overview of the enormous morphological, taxonomical and functional diversity of soil protists, and discuss current challenges and opportunities in soil protistology. Research in soil biology would clearly benefit from incorporating more protistology alongside the study of bacteria, fungi and animals.
The Protist Ribosomal Reference database (
Precipitation changes, warming, and N input differentially affect microbial predators in an alpine meadow: Evidence from soil phagotrophic protists
DOI:10.1016/j.soilbio.2021.108521 URL [本文引用: 1]
Emerging parasitic protists: The case of Perkinsea
DOI:10.3389/fmicb.2021.735815
URL
[本文引用: 1]
The last century has witnessed an increasing rate of new disease emergence across the world leading to permanent loss of biodiversity. Perkinsea is a microeukaryotic parasitic phylum composed of four main lineages of parasitic protists with broad host ranges. Some of them represent major ecological and economical threats because of their geographically invasive ability and pathogenicity (leading to mortality events). In marine environments, three lineages are currently described, the Parviluciferaceae, the Perkinsidae, and the Xcellidae, infecting, respectively, dinoflagellates, mollusks, and fish. In contrast, only one lineage is officially described in freshwater environments: the severe Perkinsea infectious agent infecting frog tadpoles. The advent of high-throughput sequencing methods, mainly based on 18S rRNA assays, showed that Perkinsea is far more diverse than the previously four described lineages especially in freshwater environments. Indeed, some lineages could be parasites of green microalgae, but a formal nature of the interaction needs to be explored. Hence, to date, most of the newly described aquatic clusters are only defined by their environmental sequences and are still not (yet) associated with any host. The unveiling of this microbial black box presents a multitude of research challenges to understand their ecological roles and ultimately to prevent their most negative impacts. This review summarizes the biological and ecological traits of Perkinsea—their diversity, life cycle, host preferences, pathogenicity, and highlights their diversity and ubiquity in association with a wide range of hosts.
An unexpected role for mixotrophs in the response of peatland carbon cycling to climate warming
DOI:10.1038/srep16931
PMID:26603894
[本文引用: 2]
Mixotrophic protists are increasingly recognized for their significant contribution to carbon (C) cycling. As phototrophs they contribute to photosynthetic C fixation, whilst as predators of decomposers, they indirectly influence organic matter decomposition. Despite these direct and indirect effects on the C cycle, little is known about the responses of peatland mixotrophs to climate change and the potential consequences for the peatland C cycle. With a combination of field and microcosm experiments, we show that mixotrophs in the Sphagnum bryosphere play an important role in modulating peatland C cycle responses to experimental warming. We found that five years of consecutive summer warming with peaks of +2 to +8 degrees C led to a 50% reduction in the biomass of the dominant mixotrophs, the mixotrophic testate amoebae (MTA). The biomass of other microbial groups (including decomposers) did not change, suggesting MTA to be particularly sensitive to temperature. In a microcosm experiment under controlled conditions, we then manipulated the abundance of MTA, and showed that the reported 50% reduction of MTA biomass in the field was linked to a significant reduction of net C uptake (-13%) of the entire Sphagnum bryosphere. Our findings suggest that reduced abundance of MTA with climate warming could lead to reduced peatland C fixation.
A novel parasitoid of marine dinoflagellates, Pararosarium dinoexitiosum gen. et sp. nov. (Perkinsozoa, Alveolata), showing characteristic beaded sporocytes
DOI:10.3389/fmicb.2021.748092
URL
[本文引用: 1]
The phylum Perkinsozoa is known as an exclusively parasitic group within alveolates and is widely distributed in various aquatic environments from marine to freshwater environments. Nonetheless, their morphology, life cycle, the identity of the host, and physiological characteristics remain still poorly understood. During intensive sampling along the west coast of Korea in October and November 2017, a new parasitoid, which shares several characteristics with the extant families Perkinsidae and Parviluciferaceae, was discovered and three strains of the new parasitoid were successfully established in cultures. Cross-infection experiments showed that among the examined planktonic groups, only dinoflagellates were susceptible to the new parasitoid, with infections observed in species belonging to eight genera. Even though the new parasitoid shared many morphological and developmental characteristics with other Perkinsozoan parasites, it differed from them by its densely packed trophocyte structure without a large vacuole or hyaline material during the growth stage. These characteristics are common among Parviluciferaceae members. Furthermore, through palintomic extracellular sporogenesis, it produced characteristic interconnected sporocytes resembling a string of beads. Phylogenetic analyses based on the small subunit and large subunit ribosomal DNA sequences revealed that the new parasitoid was distantly related to the family Parviluciferaceae and was more closely related to the families Perkinsidae and Xcellidae. Morphological, ultrastructural, and molecular data on the new parasitoid raised the need to erect a new family, i.e., Pararosariidae, within the phylum Perkinsozoa with Pararosarium dinoexitiosum gen. et sp. nov. as the type species. The isolation and establishment in culture of the new parasitoid outside the family Parviluciferaceae in the present study would contribute to the better understanding of the diversity of Perkinsozoan parasites and provide useful material for comparisons to other parasite species in the further study.
Resource partitioning between bacteria, fungi, and protists in the detritusphere of an agricultural soil
The flow of plant-derived carbon in soil is a key component of global carbon cycling. Conceptual models of trophic carbon fluxes in soil have assumed separate bacterial and fungal energy channels in the detritusphere, controlled by both substrate complexity and recalcitrance. However, detailed understanding of the key populations involved and niche-partitioning between them is limited. Here, a microcosm experiment was performed to trace the flow of detritusphere C from substrate analogs (glucose, cellulose) and plant biomass amendments (maize leaves, roots) in an agricultural soil. Carbon flow was traced by rRNA stable isotope probing and amplicon sequencing across three microbial kingdoms. Distinct lineages within the as well as were identified as important primary substrate consumers. A dynamic succession of primary consumers was observed especially in the cellulose treatments, but also in plant amendments over time. While intra-kingdom niche partitioning was clearly observed, distinct bacterial and fungal energy channels were not apparent. Furthermore, while the diversity of primary substrate consumers did not notably increase with substrate complexity, consumer succession and secondary trophic links to bacterivorous and fungivorous microbes resulted in increased food web complexity in the more recalcitrant substrates. This suggests that rather than substrate-defined energy channels, consumer succession as well as intra- and inter-kingdom cross-feeding should be considered as mechanisms supporting food web complexity in the detritusphere.
Moderate changes in nutrient input alter tropical microbial and protist communities and belowground linkages
DOI:10.1038/ismej.2013.209 [本文引用: 1]
Plant community change mediated heterotrophic respiration increase explains soil organic carbon loss before moderate degradation of alpine meadow
DOI:10.1002/ldr.v32.18 URL [本文引用: 1]
The influence of soil properties on the structure of bacterial and fungal communities across land-use types
DOI:10.1016/j.soilbio.2008.05.021 URL [本文引用: 1]
Changes of soil microbial community under different degraded gradients of alpine meadow
DOI:10.1016/j.agee.2016.02.020 URL [本文引用: 1]
Effect of grazing intensity on protozoan community, microbial biomass, and enzyme activity in an alpine meadow on the Tibetan Plateau
The degradation of subalpine meadows significantly changed the soil microbiome
DOI:10.1016/j.agee.2023.108470 URL [本文引用: 1]
Responses of soil fungal communities to subalpine meadow degradation in Mount Wutai
土壤真菌群落对五台山亚高山草甸退化的响应
Degradation-induced microbiome alterations may aggravate soil nutrient loss in subalpine meadows
Soil bacterial community response and nitrogen cycling variations associated with subalpine meadow degradation on the Loess Plateau, China
Community structures and diversity patterns of the soil protist communities along an altitudinal gradient in a subalpine grassland
亚高山草地土壤原生生物群落结构和多样性海拔分布格局
Response of soil ciliate community to ecological restoration of different return patterns
土壤纤毛虫群落对不同退还模式生态恢复的响应
The current research progress and prospects of cultivated and grassland soil health
耕地和草地土壤健康研究进展与展望
Protist taxonomic and functional diversity in soil, freshwater and marine ecosystems
DOI:10.1016/j.envint.2020.106262 URL [本文引用: 1]
Multiple marker parallel tag environmental DNA sequencing reveals a highly complex eukaryotic community in marine anoxic water
DOI:10.1111/mec.2010.19.issue-s1 URL [本文引用: 1]
Fertilization alters protistan consumers and parasites in crop-associated microbiomes
DOI:10.1111/1462-2920.15385
PMID:33400366
[本文引用: 2]
Crop plants carry an enormous diversity of microbiota that provide massive benefits to hosts. Protists, as the main microbial consumers and a pivotal driver of biogeochemical cycling processes, remain largely understudied in the plant microbiome. Here, we characterized the diversity and composition of protists in sorghum leaf phyllosphere, and rhizosphere and bulk soils, collected from an 8-year field experiment with multiple fertilization regimes. Phyllosphere was an important habitat for protists, dominated by Rhizaria, Alveolata and Amoebozoa. Rhizosphere and bulk soils had a significantly higher diversity of protists than the phyllosphere, and the protistan community structure significantly differed among the three plant-soil compartments. Fertilization significantly altered specific functional groups of protistan consumers and parasites. Variation partitioning models revealed that soil properties, bacteria and fungi predicted a significant proportion of the variation in the protistan communities. Changes in protists may in turn significantly alter the compositions of bacterial and fungal communities from the top-down control in food webs. Altogether, we provide novel evidence that fertilization significantly affects the functional groups of protistan consumers and parasites in crop-associated microbiomes, which have implications for the potential changes in their ecological functions under intensive agricultural managements.© 2021 Society for Applied Microbiology and John Wiley & Sons Ltd.
Changes in plant diversity, biomass and soil C, in alpine meadows at different degradation stages in the headwater region of three rivers, China
DOI:10.1002/ldr.v20:2 URL [本文引用: 1]
Reduction of microbial diversity in grassland soil is driven by long-term climate warming
DOI:10.1038/s41564-022-01147-3
PMID:35697795
[本文引用: 1]
Anthropogenic climate change threatens ecosystem functioning. Soil biodiversity is essential for maintaining the health of terrestrial systems, but how climate change affects the richness and abundance of soil microbial communities remains unresolved. We examined the effects of warming, altered precipitation and annual biomass removal on grassland soil bacterial, fungal and protistan communities over 7 years to determine how these representative climate changes impact microbial biodiversity and ecosystem functioning. We show that experimental warming and the concomitant reductions in soil moisture play a predominant role in shaping microbial biodiversity by decreasing the richness of bacteria (9.6%), fungi (14.5%) and protists (7.5%). Our results also show positive associations between microbial biodiversity and ecosystem functional processes, such as gross primary productivity and microbial biomass. We conclude that the detrimental effects of biodiversity loss might be more severe in a warmer world.© 2022. The Author(s), under exclusive licence to Springer Nature Limited.
Contrasting the relative importance of species sorting and dispersal limitation in shaping marine bacterial versus protist communities
DOI:10.1038/ismej.2017.183 URL [本文引用: 1]
Soil protist communities form a dynamic hub in the soil microbiome
DOI:10.1038/ismej.2017.171 URL [本文引用: 1]
Rhizosphere protists are key determinants of plant health
DOI:10.1186/s40168-020-00799-9
PMID:32127034
[本文引用: 1]
Plant health is intimately influenced by the rhizosphere microbiome, a complex assembly of organisms that changes markedly across plant growth. However, most rhizosphere microbiome research has focused on fractions of this microbiome, particularly bacteria and fungi. It remains unknown how other microbial components, especially key microbiome predators-protists-are linked to plant health. Here, we investigated the holistic rhizosphere microbiome including bacteria, microbial eukaryotes (fungi and protists), as well as functional microbial metabolism genes. We investigated these communities and functional genes throughout the growth of tomato plants that either developed disease symptoms or remained healthy under field conditions.We found that pathogen dynamics across plant growth is best predicted by protists. More specifically, communities of microbial-feeding phagotrophic protists differed between later healthy and diseased plants at plant establishment. The relative abundance of these phagotrophs negatively correlated with pathogen abundance across plant growth, suggesting that predator-prey interactions influence pathogen performance. Furthermore, phagotrophic protists likely shifted bacterial functioning by enhancing pathogen-suppressing secondary metabolite genes involved in mitigating pathogen success.We illustrate the importance of protists as top-down controllers of microbiome functioning linked to plant health. We propose that a holistic microbiome perspective, including bacteria and protists, provides the optimal next step in predicting plant performance. Video Abstract.
Grazing-induced microbiome alterations drive soil organic carbon turnover and productivity in meadow steppe
DOI:10.1186/s40168-018-0544-y
PMID:30236158
[本文引用: 1]
Background: Grazing is a major modulator of biodiversity and productivity in grasslands. However, our understanding of grazing-induced changes in below-ground communities, processes, and soil productivity is limited. Here, using a long-term enclosed grazing meadow steppe, we investigated the impacts of grazing on the soil organic carbon (SOC) turnover, the microbial community composition, resistance and activity under seasonal changes, and the microbial contributions to soil productivity.Results: The results demonstrated that grazing had significant impacts on soil microbial communities and ecosystem functions in meadow steppe. The highest microbial a-diversity was observed under light grazing intensity, while the highest beta-diversity was observed under moderate grazing intensity. Grazing shifted the microbial composition from fungi dominated to bacteria dominated and from slow growing to fast growing, thereby resulting in a shift from fungi-dominated food webs primarily utilizing recalcitrant SOC to bacteria-dominated food webs mainly utilizing labile SOC. Moreover, the higher fungal recalcitrant-SOC-decomposing activities and bacterial labile-SOC-decomposing activities were observed in fungi- and bacteria-dominated communities, respectively. Notably, the robustness of bacterial community and the stability of bacterial activity were associated with a-diversity, while this was not the case for the robustness of fungal community and its associated activities. Finally, we observed that microbial a diversity rather than SOC turnover rate can predict soil productivity.Conclusions: Our findings indicate the strong influence of grazing on soil microbial community, SOC turnover, and soil productivity and the important positive role of soil microbial a-diversity in steering the functions of meadow steppe ecosystems.
Wheat yield losses from pests and pathogens in China
DOI:10.1016/j.agee.2021.107821 URL [本文引用: 1]
Fertilization drives distinct biotic and abiotic factors in regulating functional groups of protists in a 5-year fertilization system
DOI:10.3389/fmicb.2022.1036362
URL
[本文引用: 2]
Protists play an important role in nutrient cycling, microbiome stability and soil fertility maintenance. However, the driving force of protistan functional groups remains poorly understood in agricultural ecosystems.
Protist communities are more sensitive to nitrogen fertilization than other microorganisms in diverse agricultural soils
DOI:10.1186/s40168-019-0647-0 [本文引用: 4]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}