Approaches used to detect and test hybridization: combining phylogenetic and population genetic analyses
Jian-Feng Mao1, *, , Yongpeng Ma2, Renchao Zhou3
1 National Engineering Laboratory for Tree Breeding, Key Laboratory of Genetics and Breeding in Forest Trees and Ornamental Plants of Ministry of Education, College of Biological Sciences and Technology, Beijing Forestry University, Beijing 1000832 Key Laboratory for Plant Diversity and Biogeography of East Asia, Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 6502013 School of Life Sciences, Sun Yat-sen University, Guangzhou 510275
Hybridization among diverging (interspecific or intraspecific) groups involves gene flow and genetic recombination. Increasingly, studies have shown that hybridization, a process of genetic exchanges, occurs widely in the divergence and unity of animals, plants, and microorganisms, and acts as an important mechanism for the formation and maintenance of biological diversity. The rapid development of high-throughput sequencing technology and the widespread application of genome-level techniques provides an unprecedented opportunity for us to further evaluate the universality and evolutionary significance of hybridization. However, selecting appropriate research techniques and strategies to detect the potential hybridization and evaluate its characteristics becomes a common question. In this review, we attempt to synthesize methods from phylogenetics and population genetics of the genomic era to provide biodiversity and evolutionary researchers a practical reference for testing hybridization.
Jian-FengMao, YongpengMa, RenchaoZhou. Approaches used to detect and test hybridization: combining phylogenetic and population genetic analyses[J]. Biodiversity Science, 2017, 25(6): 577-599 https://doi.org/10.17520/biods.2017097
杂交(hybridization)通常指真核生物不同类群间(种间或种内)经有性途径的遗传交换(genetic exchange)。杂交使不同亲本的遗传物质共存于杂交子代, 而杂交子代减数分裂过程中的同源染色体遗传交换实现了遗传物质在不同类群间的交换和重新组合。除了有性过程的杂交, 重组(recombination)经常被用来描述原核生物中不经有性过程实现的遗传物质重新组合。这种重组大量存在于病毒中。水平基因转移(lateral/horizontal gene transfer)指那些不同于杂交和重组中实现的基因由亲代垂直传递给子代的遗传交换过程。此外, 基因交流(gene flow)、渐渗(introgression)、伴随基因流的分化(divergence/isolation with gene flow/migration)和网状进化(reticulate evolution)等概念也常被用于概括不同时空、进化过程或系统发育特征的遗传交换。这些概念的形成、应用范围和侧重有差异, 但本质上都是遗传交换(Arnold, 2016)。不同于生命之树(tree of life)理论, 生命之网(web of life)理论认为生物的演化过程是一个由遗传交换构成的网状模式, 它在更普遍的意义上概括了遗传交换对生物多样性产生和维持的重要作用(Arnold, 2016; Mallet et al, 2016)。本文在认同不同遗传交换过程在进化生物学共性意义的基础上, 将技术策略综述的视角限定在对杂交(或者说狭义的杂交)的检测上, 以此避免大而不全带来的误导。
Fig. 1 Molecular genotyping technologies in the genomic era (adapted from Lemmon & Lemmon, 2013). The figure lists the features, operating procedures and data processing of each sequencing technique.
Fig. 2 The brief work-flow of phylogenetic strategy used to test hybridization (adapted from Lemmon & Lemmon, 2013). The work-flow contains multiple steps including data processing, data quality evaluation, data screening, orthologous gene identification, sequence analysis, selection of base substitution model, phylogenetic tree and phylogenetic network reconstruction. We sort out the hierarchy of recommended, suboptimal or requires validation and not recommended, in consideration of the degree of operation, the reliability of the data, whether can distinguish hybridization, incomplete lineage sorting and other factors. And we list the available softwares for each step.
Fig. 3 The current population genomic work-flow used to test hybridization. The basic process of global and local testing for the existence and mode of hybridization through data collection, data processing and analysis are presented. The data format and some of the available softwares are listed.
上面提到的主成分分析、聚类分析(比如基于structure和admixture的分析)可以用来澄清群体遗传结构, 并对杂交的存在提供全局性的线索(那些看似杂交的迹象可能由杂交以外的其他因素导致), 但它们都没有提供杂交是否存在的检验。比如, 与距离关联的分化(isolation by distance)可以产生在PCA上的梯度变异。基于structure和admixture的结果也很难做出对群体历史的推算, 因为它们没有对特定的群体历史模型进行检验, 而是简单地假定抽样群体都是从某特定群体快速辐射分化而来。Patterson等(2012)综合已有工作(Reich et al, 2009; Green et al, 2010; Durand et al, 2011; Moorjani et al, 2011), 归纳出了几个针对群体间杂交历史的检验和相应的参数, 并提供了实现有关计算的软件包(ADMIXTOOLS)。这些参数和检验包括1个三群体检验(f3-statistic, the three-population test)和2个四群体检验(D-statistics或者ABBA-BABA test, 以及f4- statistics或F4-ratio)。这里的f3-statistic和f4-statistics也被统称为F-statistics。
Linked selection and recombination rate variation drive the evolution of the genomic landscape of differentiation across the speciation continuum of Ficedula flycatchers.
DIYABC v2.0: a software to make approximate Bayesian computation inferences about population history using single nucleotide polymorphism, DNA sequence and microsatellite data.
Detection of interspecies hybridisation in Chondrichthyes: hybrids and hybrid offspring between Australian (Carcharhinus tilstoni) and common (C. limbatus) blacktip shark found in an Australian fishery.
Parallel tagged amplicon sequencing reveals major lineages and phylogenetic structure in the North American tiger salamander (Ambystoma tigrinum) species complex.
Development of highly reliable in silico SNP resource and genotyping assay from exome capture and sequencing: an example from black spruce (Picea mariana).
Phylogenetic marker development for target enrichment from transcriptome and genome skim data: the pipeline and its application in southern African Oxalis (Oxalidaceae).
A versatile and highly efficient toolkit including 102 nuclear markers for vertebrate phylogenomics, tested by resolving the higher level relationships of the caudata.
Needle morphological evidence of the homoploid hybrid origin of Pinus densata based on analysis of artificial hybrids and the putative parents, Pinus tabuliformis and Pinus yunnanensis.
... ).杂交对物种形成的另一个作用是, 它通过传递有利于适应性分化的等位基因, 进而强化种间隔离的形成, 并促进物种形成(Abbott et al, 2013). ...
Lateral gene transfer as a support for the tree of life
1
2012
... 模型初选后, 应该用恰当的检验来评判选定模型的可靠性.这方面有一系列的工具可以利用, 比如位点内或位点间重组(Mcguire et al, 1997; Kosakovsky et al, 2006; Martin et al, 2015)、各种选择(Delport et al, 2010)、杂交(Yu et al, 2011)、时序性进化速率差异(heterotachy) (Pagel & Meade, 2008)和模型总体上的配合性(Albert & Schluter, 2005; Abby et al, 2012; Ackermann et al, 2016)等.如果初选模型不适合, 那么应该及时调整选用更恰当的模型.如果位点间基因树冲突, 那么可考虑选用应对杂交或者不完全谱系分离的模型, 而不是仍然将序列合并使用, 或者把不相配合的部分数据删除.当然还是要删除那些体现强烈选择印记的、变异饱和的(saturated) (Castresana, 2000; Rodríguezezpeleta et al, 2007; Gnirke et al, 2009)和包含有缺失值(missing)或有缺失变异(deletion)的位点(Lemmon et al, 2009). ...
The hybrid origin of “Modern” humans.
2
2016
... 这个技术的实现是显而易见的.对于已有参考基因组、基因组大小合适的类群, 全基因组测序(更多的是基因组重测序, genome resequencing)应用已经很常见了, 尤其是对近缘种间或种内群体分化的研究中, 其优势更加明显.人类的1,000 Genomes计划(http://www.internationalgenome.org/)(The Genomes Project Consortium et al, 2010)和植物中拟南芥(Arabidopsis thaliana)的1,001 Genomes计划(http://1001genomes.org/) (Weigel & Mott, 2009)都为我们作了很好的示范.值得一提的是, 基因组水平的证据表明种间遗传交换参与了人(Patterson et al, 2012; Hellenthal et al, 2014; Lazaridis et al, 2014; Sankararaman et al, 2014; Ackermann et al, 2016)和拟南芥(Stenz et al, 2015; Novikova et al, 2016)的起源和分化. ...
... 模型初选后, 应该用恰当的检验来评判选定模型的可靠性.这方面有一系列的工具可以利用, 比如位点内或位点间重组(Mcguire et al, 1997; Kosakovsky et al, 2006; Martin et al, 2015)、各种选择(Delport et al, 2010)、杂交(Yu et al, 2011)、时序性进化速率差异(heterotachy) (Pagel & Meade, 2008)和模型总体上的配合性(Albert & Schluter, 2005; Abby et al, 2012; Ackermann et al, 2016)等.如果初选模型不适合, 那么应该及时调整选用更恰当的模型.如果位点间基因树冲突, 那么可考虑选用应对杂交或者不完全谱系分离的模型, 而不是仍然将序列合并使用, 或者把不相配合的部分数据删除.当然还是要删除那些体现强烈选择印记的、变异饱和的(saturated) (Castresana, 2000; Rodríguezezpeleta et al, 2007; Gnirke et al, 2009)和包含有缺失值(missing)或有缺失变异(deletion)的位点(Lemmon et al, 2009). ...
A comparison of multivariate methods for the detection of hybridization.
1982
Selection and the origin of species.
1
2005
... 模型初选后, 应该用恰当的检验来评判选定模型的可靠性.这方面有一系列的工具可以利用, 比如位点内或位点间重组(Mcguire et al, 1997; Kosakovsky et al, 2006; Martin et al, 2015)、各种选择(Delport et al, 2010)、杂交(Yu et al, 2011)、时序性进化速率差异(heterotachy) (Pagel & Meade, 2008)和模型总体上的配合性(Albert & Schluter, 2005; Abby et al, 2012; Ackermann et al, 2016)等.如果初选模型不适合, 那么应该及时调整选用更恰当的模型.如果位点间基因树冲突, 那么可考虑选用应对杂交或者不完全谱系分离的模型, 而不是仍然将序列合并使用, 或者把不相配合的部分数据删除.当然还是要删除那些体现强烈选择印记的、变异饱和的(saturated) (Castresana, 2000; Rodríguezezpeleta et al, 2007; Gnirke et al, 2009)和包含有缺失值(missing)或有缺失变异(deletion)的位点(Lemmon et al, 2009). ...
Direct selection of human genomic loci by microarray hybridization.
1
2007
... 目标富集测序(targeted enrichment sequencing)也称为序列捕获测序(sequence capture), 是通过高通量办法对基因组中特定序列进行富集, 进而进行高通量测序的一类技术(Mamanova et al, 2010).在目标富集测序中, 先将基因组DNA打碎, 然后通过固相(Albert et al, 2007; Hodges et al, 2007)或液相(Gnirke et al, 2009; Maricic et al, 2010)的DNA探针杂交, 将非目的片段洗脱, 捕获目的序列, 再进行样本混合建库和高通量测序.与基于限制性酶切的简化策略相比, 这个技术是有目的地对基因组中特定序列进行筛选、富集、测序.与PCR扩增产物测序相比, 目标富集测序的富集是通过探针杂交实现的, 它的捕获通量可以更高.这个技术需要提前知道要富集的序列信息(可以是基因组、转录组或其他来源的序列), 以此设计捕获探针, 实现捕获.在捕获过程中, 用于区分样本的标签可以在捕获前加入, 也可在捕获后加入(Kenny, 2011).由于捕获试剂往往较为昂贵, 捕获前加样本标签更为节省.该分子标记技术的特征参见图1. ...
Fast model-based estimation of ancestry in unrelated individuals.
1
2009
... 这里提到的聚类方法(clustering methods)是指按等位基因频率分布模式, 将个体的来源归入相应祖先群体(ancestral populations)中.简单地说, 聚类的基本过程是: 先假定有K个可能存在的祖先群体; 然后依据个体基因型计算它们被判别为各群体的概率; 在计算时, 假定各位点群体上的频率分布服从哈代-温伯格平衡(Hardy-Weinberg equilibrium).利用structure (Pritchard et al, 2000; Falush et al, 2003)和admixture (Alexander et al, 2009)等基于模型的聚类方法对每一个参试个体估算其来自于K个祖先群体中某个特定群体的遗传比例.祖先群体的数量K是个先验值, 可以依据一些已知的研究背景确定, 也可以通过最小K值的策略估算出来.旨在揭示杂交的群体遗传研究中, 此类聚类方法的应用非常普遍.InStruct是structure软件的拓展, 它没有哈代-温伯格平衡的前提假设, 可以同时对群体结构和近交率进行估算(Gao et al, 2007), 这对近交或自交比较频繁的植物研究有利用价值.类似的聚类运算的软件还有不少, 例如FRAPPE (Tang et al, 2007)、sNMF (Frichot et al, 2014)、Geneland (Guillot et al, 2004, 2005)等.它们在操作上类似, 运算速度相差不少, 有些可以考虑样本群体间的环境或地理信息.这类聚类方法可以应用于基因型数据、二倍化的单倍型数据(pseudo-homozygous genotype)或者基因型的似然值数据(genotype likelihood).ANGSD软件包中的NGSAdmix软件(Skotte et al, 2013)可以基于基因组水平基因型的似然值数据而不是基因型数据做聚类分析. ...
Hybridization in Tradescantia. III. The evidence for introgressive hybridization.
1
1938
... 越来越多的证据表明, 杂交普遍存在于生物类群分化和维持中, 具有重要的进化生物学意义.在农林业上, 杂交是一种重要的育种手段.通过人工杂交, 可以快速获得杂种优势、实现有益变异在不同育种材料间的重新组合, 甚至产生全新的表型.在自然类群中, 杂交也有类似的作用: 可以带来新的遗传变异, 且速度比突变要快得多(Anderson & Hubricht, 1938; Martinsen et al, 2001).因此, 杂交增加了选择中性位点的等位基因数量; 引入具有选择优势的等位基因, 增加获得这些变异的类群的适合度, 促成快速的适应性转变(Choler et al, 2004; Martin et al, 2006; Castric et al, 2008; Kim et al, 2008). ...
Harnessing the power of RADseq for ecological and evolutionary genomics.
1
2016
... 这类技术都包含一个重要步骤, 即对目标样本基因组进行限制性内切酶酶切, 有些还包含对酶切产物片段大小的筛选(Baird et al, 2008; van Tassell et al, 2008; Kim et al, 2016).酶切过程中, 可用单一的内切酶, 也可用两个内切酶的组合(Peterson et al, 2012); 酶切的特征可能是甲基化敏感的、甲基化不敏感的或IIB型限制性核酸内切酶(Wang et al, 2012).酶的组合和酶切片段筛选给了这类技术极大的灵活性, 加上样本标签后可以实现各种组合的混样测序, 这类技术的潜力巨大.基于类似的简化原理, 已有大量特定技术衍生出来(如reduced-representation library sequencing (RRL), restriction-site- associated DNA sequencing (RAD), genotyping by sequencing (GBS)), 有不少综述性论文对它们进行了详细介绍(Davey et al, 2011; Andrews et al, 2016).该分子标记技术的特征参见图1. ...
2
2016
... 杂交(hybridization)通常指真核生物不同类群间(种间或种内)经有性途径的遗传交换(genetic exchange).杂交使不同亲本的遗传物质共存于杂交子代, 而杂交子代减数分裂过程中的同源染色体遗传交换实现了遗传物质在不同类群间的交换和重新组合.除了有性过程的杂交, 重组(recombination)经常被用来描述原核生物中不经有性过程实现的遗传物质重新组合.这种重组大量存在于病毒中.水平基因转移(lateral/horizontal gene transfer)指那些不同于杂交和重组中实现的基因由亲代垂直传递给子代的遗传交换过程.此外, 基因交流(gene flow)、渐渗(introgression)、伴随基因流的分化(divergence/isolation with gene flow/migration)和网状进化(reticulate evolution)等概念也常被用于概括不同时空、进化过程或系统发育特征的遗传交换.这些概念的形成、应用范围和侧重有差异, 但本质上都是遗传交换(Arnold, 2016).不同于生命之树(tree of life)理论, 生命之网(web of life)理论认为生物的演化过程是一个由遗传交换构成的网状模式, 它在更普遍的意义上概括了遗传交换对生物多样性产生和维持的重要作用(Arnold, 2016; Mallet et al, 2016).本文在认同不同遗传交换过程在进化生物学共性意义的基础上, 将技术策略综述的视角限定在对杂交(或者说狭义的杂交)的检测上, 以此避免大而不全带来的误导. ...
... ).不同于生命之树(tree of life)理论, 生命之网(web of life)理论认为生物的演化过程是一个由遗传交换构成的网状模式, 它在更普遍的意义上概括了遗传交换对生物多样性产生和维持的重要作用(Arnold, 2016; Mallet et al, 2016).本文在认同不同遗传交换过程在进化生物学共性意义的基础上, 将技术策略综述的视角限定在对杂交(或者说狭义的杂交)的检测上, 以此避免大而不全带来的误导. ...
Rapid SNP discovery and genetic mapping using sequenced RAD markers.
1
2008
... 这类技术都包含一个重要步骤, 即对目标样本基因组进行限制性内切酶酶切, 有些还包含对酶切产物片段大小的筛选(Baird et al, 2008; van Tassell et al, 2008; Kim et al, 2016).酶切过程中, 可用单一的内切酶, 也可用两个内切酶的组合(Peterson et al, 2012); 酶切的特征可能是甲基化敏感的、甲基化不敏感的或IIB型限制性核酸内切酶(Wang et al, 2012).酶的组合和酶切片段筛选给了这类技术极大的灵活性, 加上样本标签后可以实现各种组合的混样测序, 这类技术的潜力巨大.基于类似的简化原理, 已有大量特定技术衍生出来(如reduced-representation library sequencing (RRL), restriction-site- associated DNA sequencing (RAD), genotyping by sequencing (GBS)), 有不少综述性论文对它们进行了详细介绍(Davey et al, 2011; Andrews et al, 2016).该分子标记技术的特征参见图1. ...
Fast and accurate inference of local ancestry in Latino populations.
1
2012
... 已经有一些估算位点祖先状态的计算工具, Padhukasahasram (2014)就进行了较为详尽的综述.一般来说, 这些计算工具需要借助隐马尔科夫模型(hidden Markov models, HMMs)来确定不同亲本来源的基因组区段的界限.HAPMIX软件可以利用未定相(unphased)的基因型数据确定杂种个体特定位点的祖先来源(Price et al, 2009).CRFs应用一个广义隐马尔科夫模型(generalized hidden Markov model), 借助训练集数据实现了对祖先来源的估算(Sankararaman et al, 2012).实现这类分析的软件还包括: SABER (Tang et al, 2006)、HAPA (Sundquist et al, 2008)、LAMP (Sankararaman et al, 2008)、LAMP- LD/LAMP-HAP (Baran et al, 2012)、WINPOP (Pasaniuc et al, 2009)、SupportMix (Omberg et al, 2012)、ASPCA (Moreno-Estrada et al, 2013)、ALLOY (Rodriguez et al, 2013)、RFMix (Maples et al, 2013)、Lanc- CSV (Brown & Pasaniuc, 2014)和EILA (Yang et al, 2013).EILA没有对输入数据位点间连锁平衡的前提假设, 且比LAMP和HAPMIX更准确, 运行速度也不慢.Lanc-CSV实现了经典算法, 但对超大样本数据有速度上的优势. ...
Enriching the ant tree of life: enhanced UCE bait set for genome-scale phylogenetics of ants and other Hymenoptera.
1
2017
... 尽管目标富集测序技术相对成熟, 但目前的应用较多地针对人类疾病研究, 在系统发育分析中的应用仍在发展.探针开发是一个重要环节, 从转录组或基因组开发探针的工具和流程已有不少报道(Mayer et al, 2016; Pavy et al, 2016; Schmickl et al, 2016).超保守序列(ultraconserved elements)也是开发捕获探针的重要来源, 目前哺乳动物(Bejerano et al, 2004; Reneker et al, 2012)、鸟类(Mccormack & Al, 2011)、两栖爬行类(Crawford et al, 2012)、昆虫(Branstetter et al, 2017)、高等植物(Freeling et al, 2009; Reneker et al, 2012)上的超保守序列已有报道.该技术还有一个特点, 就是通过对长目标序列多个不同区域设计探针, 可以实现对整条序列的捕获测序.这突破了目前流行的新一代测序的读长较短的限制.但这对数据分析也造成了问题, 因为目前流行的系统发育分析软件对大量长片段进行分析时计算速度往往很慢, 比如BEST (Edwards et al, 2007)和*BEST (Heled & Drummond, 2010). ...
Enhanced methods for local ancestry assignment in sequenced admixed individuals.
1
2014
... 已经有一些估算位点祖先状态的计算工具, Padhukasahasram (2014)就进行了较为详尽的综述.一般来说, 这些计算工具需要借助隐马尔科夫模型(hidden Markov models, HMMs)来确定不同亲本来源的基因组区段的界限.HAPMIX软件可以利用未定相(unphased)的基因型数据确定杂种个体特定位点的祖先来源(Price et al, 2009).CRFs应用一个广义隐马尔科夫模型(generalized hidden Markov model), 借助训练集数据实现了对祖先来源的估算(Sankararaman et al, 2012).实现这类分析的软件还包括: SABER (Tang et al, 2006)、HAPA (Sundquist et al, 2008)、LAMP (Sankararaman et al, 2008)、LAMP- LD/LAMP-HAP (Baran et al, 2012)、WINPOP (Pasaniuc et al, 2009)、SupportMix (Omberg et al, 2012)、ASPCA (Moreno-Estrada et al, 2013)、ALLOY (Rodriguez et al, 2013)、RFMix (Maples et al, 2013)、Lanc- CSV (Brown & Pasaniuc, 2014)和EILA (Yang et al, 2013).EILA没有对输入数据位点间连锁平衡的前提假设, 且比LAMP和HAPMIX更准确, 运行速度也不慢.Lanc-CSV实现了经典算法, 但对超大样本数据有速度上的优势. ...
Genetic recombination in plant-infecting messenger-sense RNA viruses: overview and research perspectives.
1
2013
... 生物类群中杂交的普遍存在获得了越来越多的认可.然而, 相对于复杂的生物多样性, 清晰描述的杂交事件仍显得非常少.实际上, 作为一个重要的进化过程和生物多样性维持机制, 杂交受到的关注还远远不够.据估算, 有25%的高等植物物种(Mallet, 2005)和10%的动物物种(Schwenk et al, 2008)间存在着种间杂交; 此外, 在真菌(Nelson, 1963; Depotter et al, 2016)、细菌(Duncan et al, 1989; Cohan & Kane, 2001; Cohan, 2002; Earl et al, 2008)、病毒(Worobey & Holmes, 1999; Bujarski, 2013; Lefeuvre & Moriones, 2015; Pérez-Losada et al, 2015; Su et al, 2016)的基因组进化中, 杂交普遍存在.以植物为例, Flora of China共记载我国3万余种植物(Wu et al, 2014), 按25%计算, 我们推测其中9,000种植物间存在杂交.同时, 日益加剧的全球气候变化和人类活动将对生物多样性的分布特征产生显著影响, 进而改变种间关系, 导致原已形成的种间生殖隔离改变或降低, 进一步增加杂交的机会(Stenz et al, 2015; Novikova et al, 2016; Vallejo- Marin & Hiscock, 2016).相较于上面的估算, 得到确认的植物杂交案例非常稀少, 我们认为可能不足1%.因此, 要全面澄清杂交产生的机制、杂交对多样性形成和维持的影响及其进化生物学意义还为时尚早.目前已经开展或正在进行的工作所覆盖的杂交事件, 估计不超过总量的1‰.检测杂交及评估其发生的范围和机制, 将成为未来生物多样性监测、管理和保护中不可缺少的组成部分.在后志书时代的今天, 检测杂交等遗传交换事件的存在, 进而揭示它对生物多样性形成和维持的重要价值, 是我们面临的重要挑战. ...
Linked selection and recombination rate variation drive the evolution of the genomic landscape of differentiation across the speciation continuum of Ficedula flycatchers.
1
2015
... 与杂交关联的是不同特征的基因组间的重组, 这种重组会带来一个嵌合的基因组, 在嵌合的基因组上不同的区域体现着不同的进化历程.识别这些位点不但对澄清种间或群体间的基因交流有意义(Green et al, 2010; Reich et al, 2010; Prufer et al, 2014), 同时也可以提供重组模式和适应性分化的信息(Kim & Rothschild, 2014).这些位点的识别对于揭示生殖隔离的遗传基础和找寻物种形成基因(speciation genes)有重要意义(Wu & Ting, 2004; Nosil & Schluter, 2011; Burri et al, 2015), 同时对建立针对濒危物种的保护策略有应用价值(der Sarkissian et al, 2015).基于基因组水平的滑动窗(sliding window)分析可以追溯不同染色体区段的祖先状态, 上面提到过的D-statistics就是一个很好的工具(Kronforst et al, 2013; Smith & Kronforst, 2013). ...
Inference of ancestral recombination graphs through topological data analysis.
Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis.
1
2000
... 模型初选后, 应该用恰当的检验来评判选定模型的可靠性.这方面有一系列的工具可以利用, 比如位点内或位点间重组(Mcguire et al, 1997; Kosakovsky et al, 2006; Martin et al, 2015)、各种选择(Delport et al, 2010)、杂交(Yu et al, 2011)、时序性进化速率差异(heterotachy) (Pagel & Meade, 2008)和模型总体上的配合性(Albert & Schluter, 2005; Abby et al, 2012; Ackermann et al, 2016)等.如果初选模型不适合, 那么应该及时调整选用更恰当的模型.如果位点间基因树冲突, 那么可考虑选用应对杂交或者不完全谱系分离的模型, 而不是仍然将序列合并使用, 或者把不相配合的部分数据删除.当然还是要删除那些体现强烈选择印记的、变异饱和的(saturated) (Castresana, 2000; Rodríguezezpeleta et al, 2007; Gnirke et al, 2009)和包含有缺失值(missing)或有缺失变异(deletion)的位点(Lemmon et al, 2009). ...
Repeated adaptive introgression at a gene under multiallelic balancing selection.
1
2008
... 越来越多的证据表明, 杂交普遍存在于生物类群分化和维持中, 具有重要的进化生物学意义.在农林业上, 杂交是一种重要的育种手段.通过人工杂交, 可以快速获得杂种优势、实现有益变异在不同育种材料间的重新组合, 甚至产生全新的表型.在自然类群中, 杂交也有类似的作用: 可以带来新的遗传变异, 且速度比突变要快得多(Anderson & Hubricht, 1938; Martinsen et al, 2001).因此, 杂交增加了选择中性位点的等位基因数量; 引入具有选择优势的等位基因, 增加获得这些变异的类群的适合度, 促成快速的适应性转变(Choler et al, 2004; Martin et al, 2006; Castric et al, 2008; Kim et al, 2008). ...
Mitochondrial genome sequences effectively reveal the phylogeny of Hylobates gibbons.
1
2010
... 同时, 对只关注少量位点但包含多个个体的项目, 扩增子测序有很大的吸引力(Griffin et al, 2011).在检测杂交存在方面, 一个典型应用实例是鲨鱼的研究.研究人员通过扩增子测序建立的分子标记技术, 检测到帝氏真鲨(Carcharchinus tilstoni)和黑边鳍真鲨(C. limbatus)间的杂交(Morgan et al, 2012).目前, 这项技术策略还被用于获取来自全线粒体基因组(Chan et al, 2010; Morin et al, 2010; Gunnarsdóttir et al, 2011)、全叶绿体基因组(Parks, 2009)、宏基因组(metagenomics)遗传变异的研究中. ...
Review of the application of modern cytogenetic methods (FISH/GISH) to the study of reticulation (polyploidy/hybridisation).
2010
Genetic introgression as a potential to widen a species’ niche: insights from alpine Carex curvula
1
2004
... 越来越多的证据表明, 杂交普遍存在于生物类群分化和维持中, 具有重要的进化生物学意义.在农林业上, 杂交是一种重要的育种手段.通过人工杂交, 可以快速获得杂种优势、实现有益变异在不同育种材料间的重新组合, 甚至产生全新的表型.在自然类群中, 杂交也有类似的作用: 可以带来新的遗传变异, 且速度比突变要快得多(Anderson & Hubricht, 1938; Martinsen et al, 2001).因此, 杂交增加了选择中性位点的等位基因数量; 引入具有选择优势的等位基因, 增加获得这些变异的类群的适合度, 促成快速的适应性转变(Choler et al, 2004; Martin et al, 2006; Castric et al, 2008; Kim et al, 2008). ...
Sexual isolation and speciation in bacteria.
1
2002
... 生物类群中杂交的普遍存在获得了越来越多的认可.然而, 相对于复杂的生物多样性, 清晰描述的杂交事件仍显得非常少.实际上, 作为一个重要的进化过程和生物多样性维持机制, 杂交受到的关注还远远不够.据估算, 有25%的高等植物物种(Mallet, 2005)和10%的动物物种(Schwenk et al, 2008)间存在着种间杂交; 此外, 在真菌(Nelson, 1963; Depotter et al, 2016)、细菌(Duncan et al, 1989; Cohan & Kane, 2001; Cohan, 2002; Earl et al, 2008)、病毒(Worobey & Holmes, 1999; Bujarski, 2013; Lefeuvre & Moriones, 2015; Pérez-Losada et al, 2015; Su et al, 2016)的基因组进化中, 杂交普遍存在.以植物为例, Flora of China共记载我国3万余种植物(Wu et al, 2014), 按25%计算, 我们推测其中9,000种植物间存在杂交.同时, 日益加剧的全球气候变化和人类活动将对生物多样性的分布特征产生显著影响, 进而改变种间关系, 导致原已形成的种间生殖隔离改变或降低, 进一步增加杂交的机会(Stenz et al, 2015; Novikova et al, 2016; Vallejo- Marin & Hiscock, 2016).相较于上面的估算, 得到确认的植物杂交案例非常稀少, 我们认为可能不足1%.因此, 要全面澄清杂交产生的机制、杂交对多样性形成和维持的影响及其进化生物学意义还为时尚早.目前已经开展或正在进行的工作所覆盖的杂交事件, 估计不超过总量的1‰.检测杂交及评估其发生的范围和机制, 将成为未来生物多样性监测、管理和保护中不可缺少的组成部分.在后志书时代的今天, 检测杂交等遗传交换事件的存在, 进而揭示它对生物多样性形成和维持的重要价值, 是我们面临的重要挑战. ...
Bacterial species and speciation.
1
2001
... 生物类群中杂交的普遍存在获得了越来越多的认可.然而, 相对于复杂的生物多样性, 清晰描述的杂交事件仍显得非常少.实际上, 作为一个重要的进化过程和生物多样性维持机制, 杂交受到的关注还远远不够.据估算, 有25%的高等植物物种(Mallet, 2005)和10%的动物物种(Schwenk et al, 2008)间存在着种间杂交; 此外, 在真菌(Nelson, 1963; Depotter et al, 2016)、细菌(Duncan et al, 1989; Cohan & Kane, 2001; Cohan, 2002; Earl et al, 2008)、病毒(Worobey & Holmes, 1999; Bujarski, 2013; Lefeuvre & Moriones, 2015; Pérez-Losada et al, 2015; Su et al, 2016)的基因组进化中, 杂交普遍存在.以植物为例, Flora of China共记载我国3万余种植物(Wu et al, 2014), 按25%计算, 我们推测其中9,000种植物间存在杂交.同时, 日益加剧的全球气候变化和人类活动将对生物多样性的分布特征产生显著影响, 进而改变种间关系, 导致原已形成的种间生殖隔离改变或降低, 进一步增加杂交的机会(Stenz et al, 2015; Novikova et al, 2016; Vallejo- Marin & Hiscock, 2016).相较于上面的估算, 得到确认的植物杂交案例非常稀少, 我们认为可能不足1%.因此, 要全面澄清杂交产生的机制、杂交对多样性形成和维持的影响及其进化生物学意义还为时尚早.目前已经开展或正在进行的工作所覆盖的杂交事件, 估计不超过总量的1‰.检测杂交及评估其发生的范围和机制, 将成为未来生物多样性监测、管理和保护中不可缺少的组成部分.在后志书时代的今天, 检测杂交等遗传交换事件的存在, 进而揭示它对生物多样性形成和维持的重要价值, 是我们面临的重要挑战. ...
DIYABC v2.0: a software to make approximate Bayesian computation inferences about population history using single nucleotide polymorphism, DNA sequence and microsatellite data.
1
2014
... 基于最大似然算法的策略往往很复杂, 需要大量的计算.近似贝叶斯算法(approximate Bayesian computation, ABC)不需要计算似然值, 它提供了一个检验各种复杂进化过程是否存在的平台(Beaumont, 2010).近似贝叶斯计算中, 首先对待检测过程依据一定的期望建立多重模拟, 然后将真实数据和模拟产生的数据进行比较, 再通过选取那些与真实数据最类似的模型来实现对待检验假设的推断.近似贝叶斯计算需要完成数据模拟、统计计算、模型比较等步骤, 目前有多种针对性的工具可以利用, 可参考一些重要综述文章(Csilléry et al, 2010; Sunnåker et al, 2013; Lintusaari et al, 2016).DIYABC (Cornuet et al, 2014)、ABCtoolbox (Wegmann et al, 2010)和abc (Csilléry et al, 2011)等是通常用来实现ABC计算的平台.利用基因组水平未定向数据和ABC算法揭示杂交事件存在的一个研究实例来自针对胡蜂(Biorhiza pallida)的工作(Robinson et al, 2014). ...
More than 1000 ultraconserved elements provide evidence that turtles are the sister group of archosaurs.
1
2012
... 尽管目标富集测序技术相对成熟, 但目前的应用较多地针对人类疾病研究, 在系统发育分析中的应用仍在发展.探针开发是一个重要环节, 从转录组或基因组开发探针的工具和流程已有不少报道(Mayer et al, 2016; Pavy et al, 2016; Schmickl et al, 2016).超保守序列(ultraconserved elements)也是开发捕获探针的重要来源, 目前哺乳动物(Bejerano et al, 2004; Reneker et al, 2012)、鸟类(Mccormack & Al, 2011)、两栖爬行类(Crawford et al, 2012)、昆虫(Branstetter et al, 2017)、高等植物(Freeling et al, 2009; Reneker et al, 2012)上的超保守序列已有报道.该技术还有一个特点, 就是通过对长目标序列多个不同区域设计探针, 可以实现对整条序列的捕获测序.这突破了目前流行的新一代测序的读长较短的限制.但这对数据分析也造成了问题, 因为目前流行的系统发育分析软件对大量长片段进行分析时计算速度往往很慢, 比如BEST (Edwards et al, 2007)和*BEST (Heled & Drummond, 2010). ...
Approximate Bayesian computation (ABC) in practice.
1
2010
... 基于最大似然算法的策略往往很复杂, 需要大量的计算.近似贝叶斯算法(approximate Bayesian computation, ABC)不需要计算似然值, 它提供了一个检验各种复杂进化过程是否存在的平台(Beaumont, 2010).近似贝叶斯计算中, 首先对待检测过程依据一定的期望建立多重模拟, 然后将真实数据和模拟产生的数据进行比较, 再通过选取那些与真实数据最类似的模型来实现对待检验假设的推断.近似贝叶斯计算需要完成数据模拟、统计计算、模型比较等步骤, 目前有多种针对性的工具可以利用, 可参考一些重要综述文章(Csilléry et al, 2010; Sunnåker et al, 2013; Lintusaari et al, 2016).DIYABC (Cornuet et al, 2014)、ABCtoolbox (Wegmann et al, 2010)和abc (Csilléry et al, 2011)等是通常用来实现ABC计算的平台.利用基因组水平未定向数据和ABC算法揭示杂交事件存在的一个研究实例来自针对胡蜂(Biorhiza pallida)的工作(Robinson et al, 2014). ...
abc: an R package for approximate Bayesian computation (ABC).
1
2011
... 基于最大似然算法的策略往往很复杂, 需要大量的计算.近似贝叶斯算法(approximate Bayesian computation, ABC)不需要计算似然值, 它提供了一个检验各种复杂进化过程是否存在的平台(Beaumont, 2010).近似贝叶斯计算中, 首先对待检测过程依据一定的期望建立多重模拟, 然后将真实数据和模拟产生的数据进行比较, 再通过选取那些与真实数据最类似的模型来实现对待检验假设的推断.近似贝叶斯计算需要完成数据模拟、统计计算、模型比较等步骤, 目前有多种针对性的工具可以利用, 可参考一些重要综述文章(Csilléry et al, 2010; Sunnåker et al, 2013; Lintusaari et al, 2016).DIYABC (Cornuet et al, 2014)、ABCtoolbox (Wegmann et al, 2010)和abc (Csilléry et al, 2011)等是通常用来实现ABC计算的平台.利用基因组水平未定向数据和ABC算法揭示杂交事件存在的一个研究实例来自针对胡蜂(Biorhiza pallida)的工作(Robinson et al, 2014). ...
Genome-wide genetic marker discovery and genotyping using next-generation sequencing.
1
2011
... 这类技术都包含一个重要步骤, 即对目标样本基因组进行限制性内切酶酶切, 有些还包含对酶切产物片段大小的筛选(Baird et al, 2008; van Tassell et al, 2008; Kim et al, 2016).酶切过程中, 可用单一的内切酶, 也可用两个内切酶的组合(Peterson et al, 2012); 酶切的特征可能是甲基化敏感的、甲基化不敏感的或IIB型限制性核酸内切酶(Wang et al, 2012).酶的组合和酶切片段筛选给了这类技术极大的灵活性, 加上样本标签后可以实现各种组合的混样测序, 这类技术的潜力巨大.基于类似的简化原理, 已有大量特定技术衍生出来(如reduced-representation library sequencing (RRL), restriction-site- associated DNA sequencing (RAD), genotyping by sequencing (GBS)), 有不少综述性论文对它们进行了详细介绍(Davey et al, 2011; Andrews et al, 2016).该分子标记技术的特征参见图1. ...
Discordance of species trees with their most likely gene trees.
Datamonkey 2010: a suite of phylogenetic analysis tools for evolutionary biology.
1
2010
... 模型初选后, 应该用恰当的检验来评判选定模型的可靠性.这方面有一系列的工具可以利用, 比如位点内或位点间重组(Mcguire et al, 1997; Kosakovsky et al, 2006; Martin et al, 2015)、各种选择(Delport et al, 2010)、杂交(Yu et al, 2011)、时序性进化速率差异(heterotachy) (Pagel & Meade, 2008)和模型总体上的配合性(Albert & Schluter, 2005; Abby et al, 2012; Ackermann et al, 2016)等.如果初选模型不适合, 那么应该及时调整选用更恰当的模型.如果位点间基因树冲突, 那么可考虑选用应对杂交或者不完全谱系分离的模型, 而不是仍然将序列合并使用, 或者把不相配合的部分数据删除.当然还是要删除那些体现强烈选择印记的、变异饱和的(saturated) (Castresana, 2000; Rodríguezezpeleta et al, 2007; Gnirke et al, 2009)和包含有缺失值(missing)或有缺失变异(deletion)的位点(Lemmon et al, 2009). ...
Interspecific hybridization impacts host range and pathogenicity of filamentous microbes.
1
2016
... 生物类群中杂交的普遍存在获得了越来越多的认可.然而, 相对于复杂的生物多样性, 清晰描述的杂交事件仍显得非常少.实际上, 作为一个重要的进化过程和生物多样性维持机制, 杂交受到的关注还远远不够.据估算, 有25%的高等植物物种(Mallet, 2005)和10%的动物物种(Schwenk et al, 2008)间存在着种间杂交; 此外, 在真菌(Nelson, 1963; Depotter et al, 2016)、细菌(Duncan et al, 1989; Cohan & Kane, 2001; Cohan, 2002; Earl et al, 2008)、病毒(Worobey & Holmes, 1999; Bujarski, 2013; Lefeuvre & Moriones, 2015; Pérez-Losada et al, 2015; Su et al, 2016)的基因组进化中, 杂交普遍存在.以植物为例, Flora of China共记载我国3万余种植物(Wu et al, 2014), 按25%计算, 我们推测其中9,000种植物间存在杂交.同时, 日益加剧的全球气候变化和人类活动将对生物多样性的分布特征产生显著影响, 进而改变种间关系, 导致原已形成的种间生殖隔离改变或降低, 进一步增加杂交的机会(Stenz et al, 2015; Novikova et al, 2016; Vallejo- Marin & Hiscock, 2016).相较于上面的估算, 得到确认的植物杂交案例非常稀少, 我们认为可能不足1%.因此, 要全面澄清杂交产生的机制、杂交对多样性形成和维持的影响及其进化生物学意义还为时尚早.目前已经开展或正在进行的工作所覆盖的杂交事件, 估计不超过总量的1‰.检测杂交及评估其发生的范围和机制, 将成为未来生物多样性监测、管理和保护中不可缺少的组成部分.在后志书时代的今天, 检测杂交等遗传交换事件的存在, 进而揭示它对生物多样性形成和维持的重要价值, 是我们面临的重要挑战. ...
A framework for variation discovery and genotyping using next-generation DNA sequencing data.
1
2011
... 相对传统技术来说, 这些分子标记技术的优势十分明显, 尽管数据处理中的某些具体环节还在发展, 但成熟的技术流程和相应的软件并不难获得.测序仪输出的原始数据转换、短读(short read)长数据质控、短读长数据的比对、转录组或基因组的组装、单碱基突变或单倍型获取、各步骤质量评估和数据筛选等重要环节的分析对一般的系统发育和群体遗传学的研究团队并不难实现, 有大量的高质量技术指南可以参考(DePristo et al, 2011; Nielsen et al, 2011).对某一种特定分子标记技术的分析策略也有不少参考, 比如RAD测序方面(Hapke & Thiele, 2016; Kim et al, 2016; McKinney et al, 2016; Shafer et al, 2016; Torkamaneh et al, 2016, 2017). ...
Evolutionary genomics and conservation of the endangered Przewalski’s horse.
1
2015
... 与杂交关联的是不同特征的基因组间的重组, 这种重组会带来一个嵌合的基因组, 在嵌合的基因组上不同的区域体现着不同的进化历程.识别这些位点不但对澄清种间或群体间的基因交流有意义(Green et al, 2010; Reich et al, 2010; Prufer et al, 2014), 同时也可以提供重组模式和适应性分化的信息(Kim & Rothschild, 2014).这些位点的识别对于揭示生殖隔离的遗传基础和找寻物种形成基因(speciation genes)有重要意义(Wu & Ting, 2004; Nosil & Schluter, 2011; Burri et al, 2015), 同时对建立针对濒危物种的保护策略有应用价值(der Sarkissian et al, 2015).基于基因组水平的滑动窗(sliding window)分析可以追溯不同染色体区段的祖先状态, 上面提到过的D-statistics就是一个很好的工具(Kronforst et al, 2013; Smith & Kronforst, 2013). ...
Genetic exchange between Bacillus subtilis and Bacillus licheniformis: variable hybrid stability and the nature of bacterial species.
1
1989
... 生物类群中杂交的普遍存在获得了越来越多的认可.然而, 相对于复杂的生物多样性, 清晰描述的杂交事件仍显得非常少.实际上, 作为一个重要的进化过程和生物多样性维持机制, 杂交受到的关注还远远不够.据估算, 有25%的高等植物物种(Mallet, 2005)和10%的动物物种(Schwenk et al, 2008)间存在着种间杂交; 此外, 在真菌(Nelson, 1963; Depotter et al, 2016)、细菌(Duncan et al, 1989; Cohan & Kane, 2001; Cohan, 2002; Earl et al, 2008)、病毒(Worobey & Holmes, 1999; Bujarski, 2013; Lefeuvre & Moriones, 2015; Pérez-Losada et al, 2015; Su et al, 2016)的基因组进化中, 杂交普遍存在.以植物为例, Flora of China共记载我国3万余种植物(Wu et al, 2014), 按25%计算, 我们推测其中9,000种植物间存在杂交.同时, 日益加剧的全球气候变化和人类活动将对生物多样性的分布特征产生显著影响, 进而改变种间关系, 导致原已形成的种间生殖隔离改变或降低, 进一步增加杂交的机会(Stenz et al, 2015; Novikova et al, 2016; Vallejo- Marin & Hiscock, 2016).相较于上面的估算, 得到确认的植物杂交案例非常稀少, 我们认为可能不足1%.因此, 要全面澄清杂交产生的机制、杂交对多样性形成和维持的影响及其进化生物学意义还为时尚早.目前已经开展或正在进行的工作所覆盖的杂交事件, 估计不超过总量的1‰.检测杂交及评估其发生的范围和机制, 将成为未来生物多样性监测、管理和保护中不可缺少的组成部分.在后志书时代的今天, 检测杂交等遗传交换事件的存在, 进而揭示它对生物多样性形成和维持的重要价值, 是我们面临的重要挑战. ...
Testing for ancient admixture between closely related populations.
1
2011
... 上面提到的主成分分析、聚类分析(比如基于structure和admixture的分析)可以用来澄清群体遗传结构, 并对杂交的存在提供全局性的线索(那些看似杂交的迹象可能由杂交以外的其他因素导致), 但它们都没有提供杂交是否存在的检验.比如, 与距离关联的分化(isolation by distance)可以产生在PCA上的梯度变异.基于structure和admixture的结果也很难做出对群体历史的推算, 因为它们没有对特定的群体历史模型进行检验, 而是简单地假定抽样群体都是从某特定群体快速辐射分化而来.Patterson等(2012)综合已有工作(Reich et al, 2009; Green et al, 2010; Durand et al, 2011; Moorjani et al, 2011), 归纳出了几个针对群体间杂交历史的检验和相应的参数, 并提供了实现有关计算的软件包(ADMIXTOOLS).这些参数和检验包括1个三群体检验(f3-statistic, the three-population test)和2个四群体检验(D-statistics或者ABBA-BABA test, 以及f4- statistics或F4-ratio).这里的f3-statistic和f4-statistics也被统称为F-statistics. ...
Ecology and genomics of Bacillus subtilis.
1
2008
... 生物类群中杂交的普遍存在获得了越来越多的认可.然而, 相对于复杂的生物多样性, 清晰描述的杂交事件仍显得非常少.实际上, 作为一个重要的进化过程和生物多样性维持机制, 杂交受到的关注还远远不够.据估算, 有25%的高等植物物种(Mallet, 2005)和10%的动物物种(Schwenk et al, 2008)间存在着种间杂交; 此外, 在真菌(Nelson, 1963; Depotter et al, 2016)、细菌(Duncan et al, 1989; Cohan & Kane, 2001; Cohan, 2002; Earl et al, 2008)、病毒(Worobey & Holmes, 1999; Bujarski, 2013; Lefeuvre & Moriones, 2015; Pérez-Losada et al, 2015; Su et al, 2016)的基因组进化中, 杂交普遍存在.以植物为例, Flora of China共记载我国3万余种植物(Wu et al, 2014), 按25%计算, 我们推测其中9,000种植物间存在杂交.同时, 日益加剧的全球气候变化和人类活动将对生物多样性的分布特征产生显著影响, 进而改变种间关系, 导致原已形成的种间生殖隔离改变或降低, 进一步增加杂交的机会(Stenz et al, 2015; Novikova et al, 2016; Vallejo- Marin & Hiscock, 2016).相较于上面的估算, 得到确认的植物杂交案例非常稀少, 我们认为可能不足1%.因此, 要全面澄清杂交产生的机制、杂交对多样性形成和维持的影响及其进化生物学意义还为时尚早.目前已经开展或正在进行的工作所覆盖的杂交事件, 估计不超过总量的1‰.检测杂交及评估其发生的范围和机制, 将成为未来生物多样性监测、管理和保护中不可缺少的组成部分.在后志书时代的今天, 检测杂交等遗传交换事件的存在, 进而揭示它对生物多样性形成和维持的重要价值, 是我们面临的重要挑战. ...
HaMStR: profile hidden Markov model based search for orthologs in ESTs.
1
2009
... 转录组测序(transcriptome sequencing)或者RNA测序(RNA sequencing)是针对基因组中的表达序列进行测序, 本质上也是一种简化基因组测序技术(Marioni et al, 2008; Morin et al, 2008; Wang et al, 2009).尽管这项技术通常被用于适应性进化和生态基因组学研究, 但已有的研究实例表明, 该技术获得的转录组序列可以用于重建系统发育和检测杂交的存在(Nabholz et al, 2011; Pease et al, 2016).此外, 该技术对发掘单碱基突变、进行群体遗传研究也很有吸引力, 因为它不仅可实现对复杂基因组的简化, 而且提供了来自直接与功能相关转录序列的信息.从转录组组装序列中寻找直系同源基因的计算工具已有报道, HaMStR (Ebersberger et al, 2009)是比较不错的一个.该分子标记技术的特征参见图1. ...
Conserved noncoding sequences (CNSs) in higher plants.
1
2009
... 尽管目标富集测序技术相对成熟, 但目前的应用较多地针对人类疾病研究, 在系统发育分析中的应用仍在发展.探针开发是一个重要环节, 从转录组或基因组开发探针的工具和流程已有不少报道(Mayer et al, 2016; Pavy et al, 2016; Schmickl et al, 2016).超保守序列(ultraconserved elements)也是开发捕获探针的重要来源, 目前哺乳动物(Bejerano et al, 2004; Reneker et al, 2012)、鸟类(Mccormack & Al, 2011)、两栖爬行类(Crawford et al, 2012)、昆虫(Branstetter et al, 2017)、高等植物(Freeling et al, 2009; Reneker et al, 2012)上的超保守序列已有报道.该技术还有一个特点, 就是通过对长目标序列多个不同区域设计探针, 可以实现对整条序列的捕获测序.这突破了目前流行的新一代测序的读长较短的限制.但这对数据分析也造成了问题, 因为目前流行的系统发育分析软件对大量长片段进行分析时计算速度往往很慢, 比如BEST (Edwards et al, 2007)和*BEST (Heled & Drummond, 2010). ...
Fast and efficient estimation of individual ancestry coefficients.
1
2014
... 这里提到的聚类方法(clustering methods)是指按等位基因频率分布模式, 将个体的来源归入相应祖先群体(ancestral populations)中.简单地说, 聚类的基本过程是: 先假定有K个可能存在的祖先群体; 然后依据个体基因型计算它们被判别为各群体的概率; 在计算时, 假定各位点群体上的频率分布服从哈代-温伯格平衡(Hardy-Weinberg equilibrium).利用structure (Pritchard et al, 2000; Falush et al, 2003)和admixture (Alexander et al, 2009)等基于模型的聚类方法对每一个参试个体估算其来自于K个祖先群体中某个特定群体的遗传比例.祖先群体的数量K是个先验值, 可以依据一些已知的研究背景确定, 也可以通过最小K值的策略估算出来.旨在揭示杂交的群体遗传研究中, 此类聚类方法的应用非常普遍.InStruct是structure软件的拓展, 它没有哈代-温伯格平衡的前提假设, 可以同时对群体结构和近交率进行估算(Gao et al, 2007), 这对近交或自交比较频繁的植物研究有利用价值.类似的聚类运算的软件还有不少, 例如FRAPPE (Tang et al, 2007)、sNMF (Frichot et al, 2014)、Geneland (Guillot et al, 2004, 2005)等.它们在操作上类似, 运算速度相差不少, 有些可以考虑样本群体间的环境或地理信息.这类聚类方法可以应用于基因型数据、二倍化的单倍型数据(pseudo-homozygous genotype)或者基因型的似然值数据(genotype likelihood).ANGSD软件包中的NGSAdmix软件(Skotte et al, 2013)可以基于基因组水平基因型的似然值数据而不是基因型数据做聚类分析. ...
The genome sequence of a 45,000-year-old modern human from western Siberia.
1
2014
... F4-ratio检验可以用来推断杂交造成的遗传重组的比例, 即使在不知道祖先群体的情况下, 也可以依据对系统发育关系的假定进行推断.四群体检验不但可以对杂交是否存在给出证据, 还可以提供基因流的方向.这些四群体检验也同样被广泛用于检测现代人类基因组中的古人类成分和相关的历史过程(Green et al, 2010; Fu et al, 2014, 2016; Prufer et al, 2014; Meyer et al, 2016).最近对非洲丽鱼的研究将该方法进一步拓展为五群体检验, 或称为f5-statistic (Meier et al, 2017), 该研究表明古老的杂交事件带来遗传变异, 成为后期适应性分化和物种形成的重要推动力. ...
The genetic history of Ice Age Europe.
1
2016
... F4-ratio检验可以用来推断杂交造成的遗传重组的比例, 即使在不知道祖先群体的情况下, 也可以依据对系统发育关系的假定进行推断.四群体检验不但可以对杂交是否存在给出证据, 还可以提供基因流的方向.这些四群体检验也同样被广泛用于检测现代人类基因组中的古人类成分和相关的历史过程(Green et al, 2010; Fu et al, 2014, 2016; Prufer et al, 2014; Meyer et al, 2016).最近对非洲丽鱼的研究将该方法进一步拓展为五群体检验, 或称为f5-statistic (Meier et al, 2017), 该研究表明古老的杂交事件带来遗传变异, 成为后期适应性分化和物种形成的重要推动力. ...
A Markov chain Monte Carlo approach for joint inference of population structure and inbreeding rates from multilocus genotype data.
1
2007
... 这里提到的聚类方法(clustering methods)是指按等位基因频率分布模式, 将个体的来源归入相应祖先群体(ancestral populations)中.简单地说, 聚类的基本过程是: 先假定有K个可能存在的祖先群体; 然后依据个体基因型计算它们被判别为各群体的概率; 在计算时, 假定各位点群体上的频率分布服从哈代-温伯格平衡(Hardy-Weinberg equilibrium).利用structure (Pritchard et al, 2000; Falush et al, 2003)和admixture (Alexander et al, 2009)等基于模型的聚类方法对每一个参试个体估算其来自于K个祖先群体中某个特定群体的遗传比例.祖先群体的数量K是个先验值, 可以依据一些已知的研究背景确定, 也可以通过最小K值的策略估算出来.旨在揭示杂交的群体遗传研究中, 此类聚类方法的应用非常普遍.InStruct是structure软件的拓展, 它没有哈代-温伯格平衡的前提假设, 可以同时对群体结构和近交率进行估算(Gao et al, 2007), 这对近交或自交比较频繁的植物研究有利用价值.类似的聚类运算的软件还有不少, 例如FRAPPE (Tang et al, 2007)、sNMF (Frichot et al, 2014)、Geneland (Guillot et al, 2004, 2005)等.它们在操作上类似, 运算速度相差不少, 有些可以考虑样本群体间的环境或地理信息.这类聚类方法可以应用于基因型数据、二倍化的单倍型数据(pseudo-homozygous genotype)或者基因型的似然值数据(genotype likelihood).ANGSD软件包中的NGSAdmix软件(Skotte et al, 2013)可以基于基因组水平基因型的似然值数据而不是基因型数据做聚类分析. ...
Demography and speciation history of the homoploid hybrid pine Pinus densata on the Tibetan Plateau.
1
2012
... 大量的调查分析表明, 和生殖隔离密切相关的性状更多地表现出超亲分离, 比如植物的环境适应性和开花物候性状、动物的生活史和行为性状(Rieseberg et al, 1999).总之, 杂种和亲本种间在表型的变异模式上有联系, 但这种联系还没有确定的模式可依.由此可见, 表型在揭示杂交是否存在或者确定亲本来源上有很多限制, 它无法提供确切的依据.但表型指标甚至仅仅是形态指标往往也能提供非常有价值的信息, 比如将它们与基因组水平的标记结合起来可以为物种的界定提供重要信息(Pyron et al, 2016).笔者亲历了一个体现形态数据重要性的实例. 在几十年前对松属(Pinus)的修订中, 我国的分类学家就依据形态特征猜测高山松(Pinus densata)要么来自云南松(P. yunnanensis)和油松(P. tabuliformis)的杂交, 要么作为祖先种分化为另外两种松树(吴中伦, 1956).之后, 研究人员开展了形态、生殖、物候、解剖、幼苗萌发、人工杂交、多重分子标记的群体遗传等多方面的研究, 已有的证据支持了前一个推测(Wang XR et al, 1990, 2001; Wang & Szmidt, 1994; Song et al, 2002, 2003; Ma et al, 2006; Mao et al, 2009; Wang BS et al, 2011; Gao et al, 2012; 张立沙等, 2012; 梁冬等, 2013; Xing et al, 2014; Zhao et al, 2014). ...
Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing.
2
2009
... 目标富集测序(targeted enrichment sequencing)也称为序列捕获测序(sequence capture), 是通过高通量办法对基因组中特定序列进行富集, 进而进行高通量测序的一类技术(Mamanova et al, 2010).在目标富集测序中, 先将基因组DNA打碎, 然后通过固相(Albert et al, 2007; Hodges et al, 2007)或液相(Gnirke et al, 2009; Maricic et al, 2010)的DNA探针杂交, 将非目的片段洗脱, 捕获目的序列, 再进行样本混合建库和高通量测序.与基于限制性酶切的简化策略相比, 这个技术是有目的地对基因组中特定序列进行筛选、富集、测序.与PCR扩增产物测序相比, 目标富集测序的富集是通过探针杂交实现的, 它的捕获通量可以更高.这个技术需要提前知道要富集的序列信息(可以是基因组、转录组或其他来源的序列), 以此设计捕获探针, 实现捕获.在捕获过程中, 用于区分样本的标签可以在捕获前加入, 也可在捕获后加入(Kenny, 2011).由于捕获试剂往往较为昂贵, 捕获前加样本标签更为节省.该分子标记技术的特征参见图1. ...
... 模型初选后, 应该用恰当的检验来评判选定模型的可靠性.这方面有一系列的工具可以利用, 比如位点内或位点间重组(Mcguire et al, 1997; Kosakovsky et al, 2006; Martin et al, 2015)、各种选择(Delport et al, 2010)、杂交(Yu et al, 2011)、时序性进化速率差异(heterotachy) (Pagel & Meade, 2008)和模型总体上的配合性(Albert & Schluter, 2005; Abby et al, 2012; Ackermann et al, 2016)等.如果初选模型不适合, 那么应该及时调整选用更恰当的模型.如果位点间基因树冲突, 那么可考虑选用应对杂交或者不完全谱系分离的模型, 而不是仍然将序列合并使用, 或者把不相配合的部分数据删除.当然还是要删除那些体现强烈选择印记的、变异饱和的(saturated) (Castresana, 2000; Rodríguezezpeleta et al, 2007; Gnirke et al, 2009)和包含有缺失值(missing)或有缺失变异(deletion)的位点(Lemmon et al, 2009). ...
Making next-generation sequencing work for you: approaches and practical considerations for marker development and phylogenetics.
... 上面提到的主成分分析、聚类分析(比如基于structure和admixture的分析)可以用来澄清群体遗传结构, 并对杂交的存在提供全局性的线索(那些看似杂交的迹象可能由杂交以外的其他因素导致), 但它们都没有提供杂交是否存在的检验.比如, 与距离关联的分化(isolation by distance)可以产生在PCA上的梯度变异.基于structure和admixture的结果也很难做出对群体历史的推算, 因为它们没有对特定的群体历史模型进行检验, 而是简单地假定抽样群体都是从某特定群体快速辐射分化而来.Patterson等(2012)综合已有工作(Reich et al, 2009; Green et al, 2010; Durand et al, 2011; Moorjani et al, 2011), 归纳出了几个针对群体间杂交历史的检验和相应的参数, 并提供了实现有关计算的软件包(ADMIXTOOLS).这些参数和检验包括1个三群体检验(f3-statistic, the three-population test)和2个四群体检验(D-statistics或者ABBA-BABA test, 以及f4- statistics或F4-ratio).这里的f3-statistic和f4-statistics也被统称为F-statistics. ...
... F4-ratio检验可以用来推断杂交造成的遗传重组的比例, 即使在不知道祖先群体的情况下, 也可以依据对系统发育关系的假定进行推断.四群体检验不但可以对杂交是否存在给出证据, 还可以提供基因流的方向.这些四群体检验也同样被广泛用于检测现代人类基因组中的古人类成分和相关的历史过程(Green et al, 2010; Fu et al, 2014, 2016; Prufer et al, 2014; Meyer et al, 2016).最近对非洲丽鱼的研究将该方法进一步拓展为五群体检验, 或称为f5-statistic (Meier et al, 2017), 该研究表明古老的杂交事件带来遗传变异, 成为后期适应性分化和物种形成的重要推动力. ...
... 与杂交关联的是不同特征的基因组间的重组, 这种重组会带来一个嵌合的基因组, 在嵌合的基因组上不同的区域体现着不同的进化历程.识别这些位点不但对澄清种间或群体间的基因交流有意义(Green et al, 2010; Reich et al, 2010; Prufer et al, 2014), 同时也可以提供重组模式和适应性分化的信息(Kim & Rothschild, 2014).这些位点的识别对于揭示生殖隔离的遗传基础和找寻物种形成基因(speciation genes)有重要意义(Wu & Ting, 2004; Nosil & Schluter, 2011; Burri et al, 2015), 同时对建立针对濒危物种的保护策略有应用价值(der Sarkissian et al, 2015).基于基因组水平的滑动窗(sliding window)分析可以追溯不同染色体区段的祖先状态, 上面提到过的D-statistics就是一个很好的工具(Kronforst et al, 2013; Smith & Kronforst, 2013). ...
A next-generation sequencing method for overcoming the multiple gene copy problem in polyploid phylogenetics, applied to Poa grasses.
1
2011
... 同时, 对只关注少量位点但包含多个个体的项目, 扩增子测序有很大的吸引力(Griffin et al, 2011).在检测杂交存在方面, 一个典型应用实例是鲨鱼的研究.研究人员通过扩增子测序建立的分子标记技术, 检测到帝氏真鲨(Carcharchinus tilstoni)和黑边鳍真鲨(C. limbatus)间的杂交(Morgan et al, 2012).目前, 这项技术策略还被用于获取来自全线粒体基因组(Chan et al, 2010; Morin et al, 2010; Gunnarsdóttir et al, 2011)、全叶绿体基因组(Parks, 2009)、宏基因组(metagenomics)遗传变异的研究中. ...
The ecological genetics of homoploid hybrid speciation.
... 这里提到的聚类方法(clustering methods)是指按等位基因频率分布模式, 将个体的来源归入相应祖先群体(ancestral populations)中.简单地说, 聚类的基本过程是: 先假定有K个可能存在的祖先群体; 然后依据个体基因型计算它们被判别为各群体的概率; 在计算时, 假定各位点群体上的频率分布服从哈代-温伯格平衡(Hardy-Weinberg equilibrium).利用structure (Pritchard et al, 2000; Falush et al, 2003)和admixture (Alexander et al, 2009)等基于模型的聚类方法对每一个参试个体估算其来自于K个祖先群体中某个特定群体的遗传比例.祖先群体的数量K是个先验值, 可以依据一些已知的研究背景确定, 也可以通过最小K值的策略估算出来.旨在揭示杂交的群体遗传研究中, 此类聚类方法的应用非常普遍.InStruct是structure软件的拓展, 它没有哈代-温伯格平衡的前提假设, 可以同时对群体结构和近交率进行估算(Gao et al, 2007), 这对近交或自交比较频繁的植物研究有利用价值.类似的聚类运算的软件还有不少, 例如FRAPPE (Tang et al, 2007)、sNMF (Frichot et al, 2014)、Geneland (Guillot et al, 2004, 2005)等.它们在操作上类似, 运算速度相差不少, 有些可以考虑样本群体间的环境或地理信息.这类聚类方法可以应用于基因型数据、二倍化的单倍型数据(pseudo-homozygous genotype)或者基因型的似然值数据(genotype likelihood).ANGSD软件包中的NGSAdmix软件(Skotte et al, 2013)可以基于基因组水平基因型的似然值数据而不是基因型数据做聚类分析. ...
A spatial satistical model for landscape genetics.
1
2005
... 这里提到的聚类方法(clustering methods)是指按等位基因频率分布模式, 将个体的来源归入相应祖先群体(ancestral populations)中.简单地说, 聚类的基本过程是: 先假定有K个可能存在的祖先群体; 然后依据个体基因型计算它们被判别为各群体的概率; 在计算时, 假定各位点群体上的频率分布服从哈代-温伯格平衡(Hardy-Weinberg equilibrium).利用structure (Pritchard et al, 2000; Falush et al, 2003)和admixture (Alexander et al, 2009)等基于模型的聚类方法对每一个参试个体估算其来自于K个祖先群体中某个特定群体的遗传比例.祖先群体的数量K是个先验值, 可以依据一些已知的研究背景确定, 也可以通过最小K值的策略估算出来.旨在揭示杂交的群体遗传研究中, 此类聚类方法的应用非常普遍.InStruct是structure软件的拓展, 它没有哈代-温伯格平衡的前提假设, 可以同时对群体结构和近交率进行估算(Gao et al, 2007), 这对近交或自交比较频繁的植物研究有利用价值.类似的聚类运算的软件还有不少, 例如FRAPPE (Tang et al, 2007)、sNMF (Frichot et al, 2014)、Geneland (Guillot et al, 2004, 2005)等.它们在操作上类似, 运算速度相差不少, 有些可以考虑样本群体间的环境或地理信息.这类聚类方法可以应用于基因型数据、二倍化的单倍型数据(pseudo-homozygous genotype)或者基因型的似然值数据(genotype likelihood).ANGSD软件包中的NGSAdmix软件(Skotte et al, 2013)可以基于基因组水平基因型的似然值数据而不是基因型数据做聚类分析. ...
High-throughput sequencing of complete human mtDNA genomes from the Philippines.
1
2011
... 同时, 对只关注少量位点但包含多个个体的项目, 扩增子测序有很大的吸引力(Griffin et al, 2011).在检测杂交存在方面, 一个典型应用实例是鲨鱼的研究.研究人员通过扩增子测序建立的分子标记技术, 检测到帝氏真鲨(Carcharchinus tilstoni)和黑边鳍真鲨(C. limbatus)间的杂交(Morgan et al, 2012).目前, 这项技术策略还被用于获取来自全线粒体基因组(Chan et al, 2010; Morin et al, 2010; Gunnarsdóttir et al, 2011)、全叶绿体基因组(Parks, 2009)、宏基因组(metagenomics)遗传变异的研究中. ...
Massive migration from the steppe was a source for Indo-European languages in Europe.
1
2015
... 三群体检验基于对群体间等位基因频率关联性进行评估, 可以对群体间即便是非常近期发生的杂交事件给出清晰的验证.研究人员成功地利用f3-statistic估算出尼安德特人(Homo neanderthalensis)贡献了非洲现代人群1.5-2.1%的遗传变异(Prufer et al, 2014).通过选用外类群, f3-statistic也被用来揭示多个不同类群群体的多重起源历史(Haak et al, 2015; Haber et al, 2016; Mörseburg et al, 2016). ...
Genetic evidence for an origin of the Armenians from Bronze Age mixing of multiple populations.
1
2016
... 三群体检验基于对群体间等位基因频率关联性进行评估, 可以对群体间即便是非常近期发生的杂交事件给出清晰的验证.研究人员成功地利用f3-statistic估算出尼安德特人(Homo neanderthalensis)贡献了非洲现代人群1.5-2.1%的遗传变异(Prufer et al, 2014).通过选用外类群, f3-statistic也被用来揭示多个不同类群群体的多重起源历史(Haak et al, 2015; Haber et al, 2016; Mörseburg et al, 2016). ...
GIbPSs: a toolkit for fast and accurate analyses of genotyping-by-sequencing data without a reference genome.
1
2016
... 相对传统技术来说, 这些分子标记技术的优势十分明显, 尽管数据处理中的某些具体环节还在发展, 但成熟的技术流程和相应的软件并不难获得.测序仪输出的原始数据转换、短读(short read)长数据质控、短读长数据的比对、转录组或基因组的组装、单碱基突变或单倍型获取、各步骤质量评估和数据筛选等重要环节的分析对一般的系统发育和群体遗传学的研究团队并不难实现, 有大量的高质量技术指南可以参考(DePristo et al, 2011; Nielsen et al, 2011).对某一种特定分子标记技术的分析策略也有不少参考, 比如RAD测序方面(Hapke & Thiele, 2016; Kim et al, 2016; McKinney et al, 2016; Shafer et al, 2016; Torkamaneh et al, 2016, 2017). ...
Genomic clues to the evolutionary success of polyploid plants.
adegenet: a R package for the multivariate analysis of genetic markers.
1
2008
... 主成分分析(principal component analysis, PCA)是一类经典的多元统计技术, 很早就开始被应用在遗传数据分析中(Menozzi et al, 1978).主成分分析可以将样本间的关系呈现在主成分形成的二维或三维空间上, 但它不能提供杂交存在与否的检验(Patterson et al, 2012), 介于类群间的样本不一定就来自于类群间的杂交(Yang et al, 2012).因此, EIGENSTRAT等软件基于PCA来估算杂交(Price et al, 2006)可能会得出错误的结果.除了EIGENSTRAT, adegenet也是一个主成分分析的工具(Jombart, 2008). ...
Statistical inference of allopolyploid species networks in the presence of incomplete lineage sorting.
1
2013
... 不同于上面的系统发育网络技术, 如果研究体系中只包含2个二倍体类群和它们1次杂交事件产生的1个异源四倍体类群, 并且异源四倍体中2个二倍体基因组间没有重组, 那么Allopolyploids软件中的算法可以用来在排除不完全谱系分选的情况下估算这种杂交成种的历史(https://sites.google.com/ site/touchingthedata/software/allopolyploids/)(Jones et al, 2013). ...
Multiplex target enrichment using DNA indexing for ultra-high throughput SNP Detection.
1
2011
... 目标富集测序(targeted enrichment sequencing)也称为序列捕获测序(sequence capture), 是通过高通量办法对基因组中特定序列进行富集, 进而进行高通量测序的一类技术(Mamanova et al, 2010).在目标富集测序中, 先将基因组DNA打碎, 然后通过固相(Albert et al, 2007; Hodges et al, 2007)或液相(Gnirke et al, 2009; Maricic et al, 2010)的DNA探针杂交, 将非目的片段洗脱, 捕获目的序列, 再进行样本混合建库和高通量测序.与基于限制性酶切的简化策略相比, 这个技术是有目的地对基因组中特定序列进行筛选、富集、测序.与PCR扩增产物测序相比, 目标富集测序的富集是通过探针杂交实现的, 它的捕获通量可以更高.这个技术需要提前知道要富集的序列信息(可以是基因组、转录组或其他来源的序列), 以此设计捕获探针, 实现捕获.在捕获过程中, 用于区分样本的标签可以在捕获前加入, 也可在捕获后加入(Kenny, 2011).由于捕获试剂往往较为昂贵, 捕获前加样本标签更为节省.该分子标记技术的特征参见图1. ...
Application of genotyping by sequencing technology to a variety of crop breeding programs.
2
2016
... 这类技术都包含一个重要步骤, 即对目标样本基因组进行限制性内切酶酶切, 有些还包含对酶切产物片段大小的筛选(Baird et al, 2008; van Tassell et al, 2008; Kim et al, 2016).酶切过程中, 可用单一的内切酶, 也可用两个内切酶的组合(Peterson et al, 2012); 酶切的特征可能是甲基化敏感的、甲基化不敏感的或IIB型限制性核酸内切酶(Wang et al, 2012).酶的组合和酶切片段筛选给了这类技术极大的灵活性, 加上样本标签后可以实现各种组合的混样测序, 这类技术的潜力巨大.基于类似的简化原理, 已有大量特定技术衍生出来(如reduced-representation library sequencing (RRL), restriction-site- associated DNA sequencing (RAD), genotyping by sequencing (GBS)), 有不少综述性论文对它们进行了详细介绍(Davey et al, 2011; Andrews et al, 2016).该分子标记技术的特征参见图1. ...
... 相对传统技术来说, 这些分子标记技术的优势十分明显, 尽管数据处理中的某些具体环节还在发展, 但成熟的技术流程和相应的软件并不难获得.测序仪输出的原始数据转换、短读(short read)长数据质控、短读长数据的比对、转录组或基因组的组装、单碱基突变或单倍型获取、各步骤质量评估和数据筛选等重要环节的分析对一般的系统发育和群体遗传学的研究团队并不难实现, 有大量的高质量技术指南可以参考(DePristo et al, 2011; Nielsen et al, 2011).对某一种特定分子标记技术的分析策略也有不少参考, 比如RAD测序方面(Hapke & Thiele, 2016; Kim et al, 2016; McKinney et al, 2016; Shafer et al, 2016; Torkamaneh et al, 2016, 2017). ...
Genomic adaptation of admixed dairy cattle in East Africa.
1
2014
... 与杂交关联的是不同特征的基因组间的重组, 这种重组会带来一个嵌合的基因组, 在嵌合的基因组上不同的区域体现着不同的进化历程.识别这些位点不但对澄清种间或群体间的基因交流有意义(Green et al, 2010; Reich et al, 2010; Prufer et al, 2014), 同时也可以提供重组模式和适应性分化的信息(Kim & Rothschild, 2014).这些位点的识别对于揭示生殖隔离的遗传基础和找寻物种形成基因(speciation genes)有重要意义(Wu & Ting, 2004; Nosil & Schluter, 2011; Burri et al, 2015), 同时对建立针对濒危物种的保护策略有应用价值(der Sarkissian et al, 2015).基于基因组水平的滑动窗(sliding window)分析可以追溯不同染色体区段的祖先状态, 上面提到过的D-statistics就是一个很好的工具(Kronforst et al, 2013; Smith & Kronforst, 2013). ...
Regulatory genes control a key morphological and ecological trait transferred between species.
1
2008
... 越来越多的证据表明, 杂交普遍存在于生物类群分化和维持中, 具有重要的进化生物学意义.在农林业上, 杂交是一种重要的育种手段.通过人工杂交, 可以快速获得杂种优势、实现有益变异在不同育种材料间的重新组合, 甚至产生全新的表型.在自然类群中, 杂交也有类似的作用: 可以带来新的遗传变异, 且速度比突变要快得多(Anderson & Hubricht, 1938; Martinsen et al, 2001).因此, 杂交增加了选择中性位点的等位基因数量; 引入具有选择优势的等位基因, 增加获得这些变异的类群的适合度, 促成快速的适应性转变(Choler et al, 2004; Martin et al, 2006; Castric et al, 2008; Kim et al, 2008). ...
GARD: a genetic algorithm for recombination detection.
1
2006
... 模型初选后, 应该用恰当的检验来评判选定模型的可靠性.这方面有一系列的工具可以利用, 比如位点内或位点间重组(Mcguire et al, 1997; Kosakovsky et al, 2006; Martin et al, 2015)、各种选择(Delport et al, 2010)、杂交(Yu et al, 2011)、时序性进化速率差异(heterotachy) (Pagel & Meade, 2008)和模型总体上的配合性(Albert & Schluter, 2005; Abby et al, 2012; Ackermann et al, 2016)等.如果初选模型不适合, 那么应该及时调整选用更恰当的模型.如果位点间基因树冲突, 那么可考虑选用应对杂交或者不完全谱系分离的模型, 而不是仍然将序列合并使用, 或者把不相配合的部分数据删除.当然还是要删除那些体现强烈选择印记的、变异饱和的(saturated) (Castresana, 2000; Rodríguezezpeleta et al, 2007; Gnirke et al, 2009)和包含有缺失值(missing)或有缺失变异(deletion)的位点(Lemmon et al, 2009). ...
Hybridization reveals the evolving genomic architecture of speciation.
1
2013
... 与杂交关联的是不同特征的基因组间的重组, 这种重组会带来一个嵌合的基因组, 在嵌合的基因组上不同的区域体现着不同的进化历程.识别这些位点不但对澄清种间或群体间的基因交流有意义(Green et al, 2010; Reich et al, 2010; Prufer et al, 2014), 同时也可以提供重组模式和适应性分化的信息(Kim & Rothschild, 2014).这些位点的识别对于揭示生殖隔离的遗传基础和找寻物种形成基因(speciation genes)有重要意义(Wu & Ting, 2004; Nosil & Schluter, 2011; Burri et al, 2015), 同时对建立针对濒危物种的保护策略有应用价值(der Sarkissian et al, 2015).基于基因组水平的滑动窗(sliding window)分析可以追溯不同染色体区段的祖先状态, 上面提到过的D-statistics就是一个很好的工具(Kronforst et al, 2013; Smith & Kronforst, 2013). ...
STEM: species tree estimation using maximum likelihood for gene trees under coalescence.
Recombination as a motor of host switches and virus emergence: geminiviruses as case studies.
1
2015
... 生物类群中杂交的普遍存在获得了越来越多的认可.然而, 相对于复杂的生物多样性, 清晰描述的杂交事件仍显得非常少.实际上, 作为一个重要的进化过程和生物多样性维持机制, 杂交受到的关注还远远不够.据估算, 有25%的高等植物物种(Mallet, 2005)和10%的动物物种(Schwenk et al, 2008)间存在着种间杂交; 此外, 在真菌(Nelson, 1963; Depotter et al, 2016)、细菌(Duncan et al, 1989; Cohan & Kane, 2001; Cohan, 2002; Earl et al, 2008)、病毒(Worobey & Holmes, 1999; Bujarski, 2013; Lefeuvre & Moriones, 2015; Pérez-Losada et al, 2015; Su et al, 2016)的基因组进化中, 杂交普遍存在.以植物为例, Flora of China共记载我国3万余种植物(Wu et al, 2014), 按25%计算, 我们推测其中9,000种植物间存在杂交.同时, 日益加剧的全球气候变化和人类活动将对生物多样性的分布特征产生显著影响, 进而改变种间关系, 导致原已形成的种间生殖隔离改变或降低, 进一步增加杂交的机会(Stenz et al, 2015; Novikova et al, 2016; Vallejo- Marin & Hiscock, 2016).相较于上面的估算, 得到确认的植物杂交案例非常稀少, 我们认为可能不足1%.因此, 要全面澄清杂交产生的机制、杂交对多样性形成和维持的影响及其进化生物学意义还为时尚早.目前已经开展或正在进行的工作所覆盖的杂交事件, 估计不超过总量的1‰.检测杂交及评估其发生的范围和机制, 将成为未来生物多样性监测、管理和保护中不可缺少的组成部分.在后志书时代的今天, 检测杂交等遗传交换事件的存在, 进而揭示它对生物多样性形成和维持的重要价值, 是我们面临的重要挑战. ...
The effect of ambiguous data on phylogenetic estimates obtained by maximum likelihood and Bayesian inference.
1
2009
... 模型初选后, 应该用恰当的检验来评判选定模型的可靠性.这方面有一系列的工具可以利用, 比如位点内或位点间重组(Mcguire et al, 1997; Kosakovsky et al, 2006; Martin et al, 2015)、各种选择(Delport et al, 2010)、杂交(Yu et al, 2011)、时序性进化速率差异(heterotachy) (Pagel & Meade, 2008)和模型总体上的配合性(Albert & Schluter, 2005; Abby et al, 2012; Ackermann et al, 2016)等.如果初选模型不适合, 那么应该及时调整选用更恰当的模型.如果位点间基因树冲突, 那么可考虑选用应对杂交或者不完全谱系分离的模型, 而不是仍然将序列合并使用, 或者把不相配合的部分数据删除.当然还是要删除那些体现强烈选择印记的、变异饱和的(saturated) (Castresana, 2000; Rodríguezezpeleta et al, 2007; Gnirke et al, 2009)和包含有缺失值(missing)或有缺失变异(deletion)的位点(Lemmon et al, 2009). ...
The importance of proper model assumption in Bayesian phylogenetics.
... Molecular genotyping technologies in the genomic era (adapted from Lemmon & Lemmon, 2013). The figure lists the features, operating procedures and data processing of each sequencing technique. ...
... The brief work-flow of phylogenetic strategy used to test hybridization (adapted from Lemmon & Lemmon, 2013). The work-flow contains multiple steps including data processing, data quality evaluation, data screening, orthologous gene identification, sequence analysis, selection of base substitution model, phylogenetic tree and phylogenetic network reconstruction. We sort out the hierarchy of recommended, suboptimal or requires validation and not recommended, in consideration of the degree of operation, the reliability of the data, whether can distinguish hybridization, incomplete lineage sorting and other factors. And we list the available softwares for each step. ...
高山松及其亲本种群在油松生境下的苗期性状
1
2013
... 大量的调查分析表明, 和生殖隔离密切相关的性状更多地表现出超亲分离, 比如植物的环境适应性和开花物候性状、动物的生活史和行为性状(Rieseberg et al, 1999).总之, 杂种和亲本种间在表型的变异模式上有联系, 但这种联系还没有确定的模式可依.由此可见, 表型在揭示杂交是否存在或者确定亲本来源上有很多限制, 它无法提供确切的依据.但表型指标甚至仅仅是形态指标往往也能提供非常有价值的信息, 比如将它们与基因组水平的标记结合起来可以为物种的界定提供重要信息(Pyron et al, 2016).笔者亲历了一个体现形态数据重要性的实例. 在几十年前对松属(Pinus)的修订中, 我国的分类学家就依据形态特征猜测高山松(Pinus densata)要么来自云南松(P. yunnanensis)和油松(P. tabuliformis)的杂交, 要么作为祖先种分化为另外两种松树(吴中伦, 1956).之后, 研究人员开展了形态、生殖、物候、解剖、幼苗萌发、人工杂交、多重分子标记的群体遗传等多方面的研究, 已有的证据支持了前一个推测(Wang XR et al, 1990, 2001; Wang & Szmidt, 1994; Song et al, 2002, 2003; Ma et al, 2006; Mao et al, 2009; Wang BS et al, 2011; Gao et al, 2012; 张立沙等, 2012; 梁冬等, 2013; Xing et al, 2014; Zhao et al, 2014). ...
高山松及其亲本种群在油松生境下的苗期性状
1
2013
... 大量的调查分析表明, 和生殖隔离密切相关的性状更多地表现出超亲分离, 比如植物的环境适应性和开花物候性状、动物的生活史和行为性状(Rieseberg et al, 1999).总之, 杂种和亲本种间在表型的变异模式上有联系, 但这种联系还没有确定的模式可依.由此可见, 表型在揭示杂交是否存在或者确定亲本来源上有很多限制, 它无法提供确切的依据.但表型指标甚至仅仅是形态指标往往也能提供非常有价值的信息, 比如将它们与基因组水平的标记结合起来可以为物种的界定提供重要信息(Pyron et al, 2016).笔者亲历了一个体现形态数据重要性的实例. 在几十年前对松属(Pinus)的修订中, 我国的分类学家就依据形态特征猜测高山松(Pinus densata)要么来自云南松(P. yunnanensis)和油松(P. tabuliformis)的杂交, 要么作为祖先种分化为另外两种松树(吴中伦, 1956).之后, 研究人员开展了形态、生殖、物候、解剖、幼苗萌发、人工杂交、多重分子标记的群体遗传等多方面的研究, 已有的证据支持了前一个推测(Wang XR et al, 1990, 2001; Wang & Szmidt, 1994; Song et al, 2002, 2003; Ma et al, 2006; Mao et al, 2009; Wang BS et al, 2011; Gao et al, 2012; 张立沙等, 2012; 梁冬等, 2013; Xing et al, 2014; Zhao et al, 2014). ...
Fundamentals and recent developments in approximate Bayesian computation.
1
2016
... 基于最大似然算法的策略往往很复杂, 需要大量的计算.近似贝叶斯算法(approximate Bayesian computation, ABC)不需要计算似然值, 它提供了一个检验各种复杂进化过程是否存在的平台(Beaumont, 2010).近似贝叶斯计算中, 首先对待检测过程依据一定的期望建立多重模拟, 然后将真实数据和模拟产生的数据进行比较, 再通过选取那些与真实数据最类似的模型来实现对待检验假设的推断.近似贝叶斯计算需要完成数据模拟、统计计算、模型比较等步骤, 目前有多种针对性的工具可以利用, 可参考一些重要综述文章(Csilléry et al, 2010; Sunnåker et al, 2013; Lintusaari et al, 2016).DIYABC (Cornuet et al, 2014)、ABCtoolbox (Wegmann et al, 2010)和abc (Csilléry et al, 2011)等是通常用来实现ABC计算的平台.利用基因组水平未定向数据和ABC算法揭示杂交事件存在的一个研究实例来自针对胡蜂(Biorhiza pallida)的工作(Robinson et al, 2014). ...
Efficient moment-based inference of admixture parameters and sources of gene flow.
1
2013
... 重组图(admixture graph)是一个允许类群间基因交流的系统发育树的拓展.基于上面提到的F-statistics就可以构建这样的图, 目前有几种工具可以利用.MixMapper (Lipson et al, 2014)是一个半自动的工具.它首先基于由一对明显没有杂交的群体间等位基因频率(F2)求算的遗传聚类生成邻接树(neighbor-joining tree), 然后在允许基因流的情况下把剩下的群体添加到图上来(Lipson et al, 2013, 2014).TreeMix (Pickrell & Pritchard, 2012)实现了与MixMapper在理论上类似的算法.在假定重组或者基因流个数的情况下, 它可以全自动地计算出重组图.不同的重组次数的假定会对结果带来极大影响, 选取时需谨慎.上面提到的ADMIXTOOLS软件(Patterson et al, 2012)中也提供了类似的工具.但这里的重组图工具更加稳健, 因为它提供了假设中的重组模式是否和数据相符的检验.重组图的手段已经在人类遗传领域广泛应用, 曾被用来揭示北美原著民的遗传组成和来源(Raghavan et al, 2014b)、丹尼索瓦人(Denisovans)对现代人群的遗传贡献(Meyer et al, 2012)、新世界寒带地区的人类定居历史(Raghavan et al, 2014a). ...
Reconstructing Austronesian population history in Island Southeast Asia.
2
2014
... 重组图(admixture graph)是一个允许类群间基因交流的系统发育树的拓展.基于上面提到的F-statistics就可以构建这样的图, 目前有几种工具可以利用.MixMapper (Lipson et al, 2014)是一个半自动的工具.它首先基于由一对明显没有杂交的群体间等位基因频率(F2)求算的遗传聚类生成邻接树(neighbor-joining tree), 然后在允许基因流的情况下把剩下的群体添加到图上来(Lipson et al, 2013, 2014).TreeMix (Pickrell & Pritchard, 2012)实现了与MixMapper在理论上类似的算法.在假定重组或者基因流个数的情况下, 它可以全自动地计算出重组图.不同的重组次数的假定会对结果带来极大影响, 选取时需谨慎.上面提到的ADMIXTOOLS软件(Patterson et al, 2012)中也提供了类似的工具.但这里的重组图工具更加稳健, 因为它提供了假设中的重组模式是否和数据相符的检验.重组图的手段已经在人类遗传领域广泛应用, 曾被用来揭示北美原著民的遗传组成和来源(Raghavan et al, 2014b)、丹尼索瓦人(Denisovans)对现代人群的遗传贡献(Meyer et al, 2012)、新世界寒带地区的人类定居历史(Raghavan et al, 2014a). ...
... , 2014).TreeMix (Pickrell & Pritchard, 2012)实现了与MixMapper在理论上类似的算法.在假定重组或者基因流个数的情况下, 它可以全自动地计算出重组图.不同的重组次数的假定会对结果带来极大影响, 选取时需谨慎.上面提到的ADMIXTOOLS软件(Patterson et al, 2012)中也提供了类似的工具.但这里的重组图工具更加稳健, 因为它提供了假设中的重组模式是否和数据相符的检验.重组图的手段已经在人类遗传领域广泛应用, 曾被用来揭示北美原著民的遗传组成和来源(Raghavan et al, 2014b)、丹尼索瓦人(Denisovans)对现代人群的遗传贡献(Meyer et al, 2012)、新世界寒带地区的人类定居历史(Raghavan et al, 2014a). ...
BEST: Bayesian estimation of species trees under the coalescent model.
Multi-layered population structure in Island Southeast Asians.
1
2016
... 三群体检验基于对群体间等位基因频率关联性进行评估, 可以对群体间即便是非常近期发生的杂交事件给出清晰的验证.研究人员成功地利用f3-statistic估算出尼安德特人(Homo neanderthalensis)贡献了非洲现代人群1.5-2.1%的遗传变异(Prufer et al, 2014).通过选用外类群, f3-statistic也被用来揭示多个不同类群群体的多重起源历史(Haak et al, 2015; Haber et al, 2016; Mörseburg et al, 2016). ...
Genetic structure and evolutionary history of a diploid hybrid pine Pinus densata inferred from the nucleotide variation at seven gene loci.
1
2006
... 大量的调查分析表明, 和生殖隔离密切相关的性状更多地表现出超亲分离, 比如植物的环境适应性和开花物候性状、动物的生活史和行为性状(Rieseberg et al, 1999).总之, 杂种和亲本种间在表型的变异模式上有联系, 但这种联系还没有确定的模式可依.由此可见, 表型在揭示杂交是否存在或者确定亲本来源上有很多限制, 它无法提供确切的依据.但表型指标甚至仅仅是形态指标往往也能提供非常有价值的信息, 比如将它们与基因组水平的标记结合起来可以为物种的界定提供重要信息(Pyron et al, 2016).笔者亲历了一个体现形态数据重要性的实例. 在几十年前对松属(Pinus)的修订中, 我国的分类学家就依据形态特征猜测高山松(Pinus densata)要么来自云南松(P. yunnanensis)和油松(P. tabuliformis)的杂交, 要么作为祖先种分化为另外两种松树(吴中伦, 1956).之后, 研究人员开展了形态、生殖、物候、解剖、幼苗萌发、人工杂交、多重分子标记的群体遗传等多方面的研究, 已有的证据支持了前一个推测(Wang XR et al, 1990, 2001; Wang & Szmidt, 1994; Song et al, 2002, 2003; Ma et al, 2006; Mao et al, 2009; Wang BS et al, 2011; Gao et al, 2012; 张立沙等, 2012; 梁冬等, 2013; Xing et al, 2014; Zhao et al, 2014). ...
Hybridization as an invasion of the genome.
1
2005
... 生物类群中杂交的普遍存在获得了越来越多的认可.然而, 相对于复杂的生物多样性, 清晰描述的杂交事件仍显得非常少.实际上, 作为一个重要的进化过程和生物多样性维持机制, 杂交受到的关注还远远不够.据估算, 有25%的高等植物物种(Mallet, 2005)和10%的动物物种(Schwenk et al, 2008)间存在着种间杂交; 此外, 在真菌(Nelson, 1963; Depotter et al, 2016)、细菌(Duncan et al, 1989; Cohan & Kane, 2001; Cohan, 2002; Earl et al, 2008)、病毒(Worobey & Holmes, 1999; Bujarski, 2013; Lefeuvre & Moriones, 2015; Pérez-Losada et al, 2015; Su et al, 2016)的基因组进化中, 杂交普遍存在.以植物为例, Flora of China共记载我国3万余种植物(Wu et al, 2014), 按25%计算, 我们推测其中9,000种植物间存在杂交.同时, 日益加剧的全球气候变化和人类活动将对生物多样性的分布特征产生显著影响, 进而改变种间关系, 导致原已形成的种间生殖隔离改变或降低, 进一步增加杂交的机会(Stenz et al, 2015; Novikova et al, 2016; Vallejo- Marin & Hiscock, 2016).相较于上面的估算, 得到确认的植物杂交案例非常稀少, 我们认为可能不足1%.因此, 要全面澄清杂交产生的机制、杂交对多样性形成和维持的影响及其进化生物学意义还为时尚早.目前已经开展或正在进行的工作所覆盖的杂交事件, 估计不超过总量的1‰.检测杂交及评估其发生的范围和机制, 将成为未来生物多样性监测、管理和保护中不可缺少的组成部分.在后志书时代的今天, 检测杂交等遗传交换事件的存在, 进而揭示它对生物多样性形成和维持的重要价值, 是我们面临的重要挑战. ...
How reticulated are species?
1
2016
... 杂交(hybridization)通常指真核生物不同类群间(种间或种内)经有性途径的遗传交换(genetic exchange).杂交使不同亲本的遗传物质共存于杂交子代, 而杂交子代减数分裂过程中的同源染色体遗传交换实现了遗传物质在不同类群间的交换和重新组合.除了有性过程的杂交, 重组(recombination)经常被用来描述原核生物中不经有性过程实现的遗传物质重新组合.这种重组大量存在于病毒中.水平基因转移(lateral/horizontal gene transfer)指那些不同于杂交和重组中实现的基因由亲代垂直传递给子代的遗传交换过程.此外, 基因交流(gene flow)、渐渗(introgression)、伴随基因流的分化(divergence/isolation with gene flow/migration)和网状进化(reticulate evolution)等概念也常被用于概括不同时空、进化过程或系统发育特征的遗传交换.这些概念的形成、应用范围和侧重有差异, 但本质上都是遗传交换(Arnold, 2016).不同于生命之树(tree of life)理论, 生命之网(web of life)理论认为生物的演化过程是一个由遗传交换构成的网状模式, 它在更普遍的意义上概括了遗传交换对生物多样性产生和维持的重要作用(Arnold, 2016; Mallet et al, 2016).本文在认同不同遗传交换过程在进化生物学共性意义的基础上, 将技术策略综述的视角限定在对杂交(或者说狭义的杂交)的检测上, 以此避免大而不全带来的误导. ...
Target-enrichment strategies for next-generation sequencing.
... 目标富集测序(targeted enrichment sequencing)也称为序列捕获测序(sequence capture), 是通过高通量办法对基因组中特定序列进行富集, 进而进行高通量测序的一类技术(Mamanova et al, 2010).在目标富集测序中, 先将基因组DNA打碎, 然后通过固相(Albert et al, 2007; Hodges et al, 2007)或液相(Gnirke et al, 2009; Maricic et al, 2010)的DNA探针杂交, 将非目的片段洗脱, 捕获目的序列, 再进行样本混合建库和高通量测序.与基于限制性酶切的简化策略相比, 这个技术是有目的地对基因组中特定序列进行筛选、富集、测序.与PCR扩增产物测序相比, 目标富集测序的富集是通过探针杂交实现的, 它的捕获通量可以更高.这个技术需要提前知道要富集的序列信息(可以是基因组、转录组或其他来源的序列), 以此设计捕获探针, 实现捕获.在捕获过程中, 用于区分样本的标签可以在捕获前加入, 也可在捕获后加入(Kenny, 2011).由于捕获试剂往往较为昂贵, 捕获前加样本标签更为节省.该分子标记技术的特征参见图1. ...
Empirical assessment of the reproductive fitness components of the hybrid pine Pinus densata on the Tibetan Plateau.
1
2009
... 大量的调查分析表明, 和生殖隔离密切相关的性状更多地表现出超亲分离, 比如植物的环境适应性和开花物候性状、动物的生活史和行为性状(Rieseberg et al, 1999).总之, 杂种和亲本种间在表型的变异模式上有联系, 但这种联系还没有确定的模式可依.由此可见, 表型在揭示杂交是否存在或者确定亲本来源上有很多限制, 它无法提供确切的依据.但表型指标甚至仅仅是形态指标往往也能提供非常有价值的信息, 比如将它们与基因组水平的标记结合起来可以为物种的界定提供重要信息(Pyron et al, 2016).笔者亲历了一个体现形态数据重要性的实例. 在几十年前对松属(Pinus)的修订中, 我国的分类学家就依据形态特征猜测高山松(Pinus densata)要么来自云南松(P. yunnanensis)和油松(P. tabuliformis)的杂交, 要么作为祖先种分化为另外两种松树(吴中伦, 1956).之后, 研究人员开展了形态、生殖、物候、解剖、幼苗萌发、人工杂交、多重分子标记的群体遗传等多方面的研究, 已有的证据支持了前一个推测(Wang XR et al, 1990, 2001; Wang & Szmidt, 1994; Song et al, 2002, 2003; Ma et al, 2006; Mao et al, 2009; Wang BS et al, 2011; Gao et al, 2012; 张立沙等, 2012; 梁冬等, 2013; Xing et al, 2014; Zhao et al, 2014). ...
RFMix: a discriminative modeling approach for rapid and robust local-ancestry inference.
1
2013
... 已经有一些估算位点祖先状态的计算工具, Padhukasahasram (2014)就进行了较为详尽的综述.一般来说, 这些计算工具需要借助隐马尔科夫模型(hidden Markov models, HMMs)来确定不同亲本来源的基因组区段的界限.HAPMIX软件可以利用未定相(unphased)的基因型数据确定杂种个体特定位点的祖先来源(Price et al, 2009).CRFs应用一个广义隐马尔科夫模型(generalized hidden Markov model), 借助训练集数据实现了对祖先来源的估算(Sankararaman et al, 2012).实现这类分析的软件还包括: SABER (Tang et al, 2006)、HAPA (Sundquist et al, 2008)、LAMP (Sankararaman et al, 2008)、LAMP- LD/LAMP-HAP (Baran et al, 2012)、WINPOP (Pasaniuc et al, 2009)、SupportMix (Omberg et al, 2012)、ASPCA (Moreno-Estrada et al, 2013)、ALLOY (Rodriguez et al, 2013)、RFMix (Maples et al, 2013)、Lanc- CSV (Brown & Pasaniuc, 2014)和EILA (Yang et al, 2013).EILA没有对输入数据位点间连锁平衡的前提假设, 且比LAMP和HAPMIX更准确, 运行速度也不慢.Lanc-CSV实现了经典算法, 但对超大样本数据有速度上的优势. ...
Why is population information crucial for taxonomy? A case study involving a hybrid swarm and related varieties.
Multiplexed DNA sequence capture of mitochondrial genomes using PCR products.
1
2010
... 目标富集测序(targeted enrichment sequencing)也称为序列捕获测序(sequence capture), 是通过高通量办法对基因组中特定序列进行富集, 进而进行高通量测序的一类技术(Mamanova et al, 2010).在目标富集测序中, 先将基因组DNA打碎, 然后通过固相(Albert et al, 2007; Hodges et al, 2007)或液相(Gnirke et al, 2009; Maricic et al, 2010)的DNA探针杂交, 将非目的片段洗脱, 捕获目的序列, 再进行样本混合建库和高通量测序.与基于限制性酶切的简化策略相比, 这个技术是有目的地对基因组中特定序列进行筛选、富集、测序.与PCR扩增产物测序相比, 目标富集测序的富集是通过探针杂交实现的, 它的捕获通量可以更高.这个技术需要提前知道要富集的序列信息(可以是基因组、转录组或其他来源的序列), 以此设计捕获探针, 实现捕获.在捕获过程中, 用于区分样本的标签可以在捕获前加入, 也可在捕获后加入(Kenny, 2011).由于捕获试剂往往较为昂贵, 捕获前加样本标签更为节省.该分子标记技术的特征参见图1. ...
RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays.
1
2008
... 转录组测序(transcriptome sequencing)或者RNA测序(RNA sequencing)是针对基因组中的表达序列进行测序, 本质上也是一种简化基因组测序技术(Marioni et al, 2008; Morin et al, 2008; Wang et al, 2009).尽管这项技术通常被用于适应性进化和生态基因组学研究, 但已有的研究实例表明, 该技术获得的转录组序列可以用于重建系统发育和检测杂交的存在(Nabholz et al, 2011; Pease et al, 2016).此外, 该技术对发掘单碱基突变、进行群体遗传研究也很有吸引力, 因为它不仅可实现对复杂基因组的简化, 而且提供了来自直接与功能相关转录序列的信息.从转录组组装序列中寻找直系同源基因的计算工具已有报道, HaMStR (Ebersberger et al, 2009)是比较不错的一个.该分子标记技术的特征参见图1. ...
RDP4: detection and analysis of recombination patterns in virus genomes.
1
2015
... 模型初选后, 应该用恰当的检验来评判选定模型的可靠性.这方面有一系列的工具可以利用, 比如位点内或位点间重组(Mcguire et al, 1997; Kosakovsky et al, 2006; Martin et al, 2015)、各种选择(Delport et al, 2010)、杂交(Yu et al, 2011)、时序性进化速率差异(heterotachy) (Pagel & Meade, 2008)和模型总体上的配合性(Albert & Schluter, 2005; Abby et al, 2012; Ackermann et al, 2016)等.如果初选模型不适合, 那么应该及时调整选用更恰当的模型.如果位点间基因树冲突, 那么可考虑选用应对杂交或者不完全谱系分离的模型, 而不是仍然将序列合并使用, 或者把不相配合的部分数据删除.当然还是要删除那些体现强烈选择印记的、变异饱和的(saturated) (Castresana, 2000; Rodríguezezpeleta et al, 2007; Gnirke et al, 2009)和包含有缺失值(missing)或有缺失变异(deletion)的位点(Lemmon et al, 2009). ...
Detecting adaptive trait introgression between Iris fulva and I. brevicaulis in highly selective field conditions.
1
2006
... 越来越多的证据表明, 杂交普遍存在于生物类群分化和维持中, 具有重要的进化生物学意义.在农林业上, 杂交是一种重要的育种手段.通过人工杂交, 可以快速获得杂种优势、实现有益变异在不同育种材料间的重新组合, 甚至产生全新的表型.在自然类群中, 杂交也有类似的作用: 可以带来新的遗传变异, 且速度比突变要快得多(Anderson & Hubricht, 1938; Martinsen et al, 2001).因此, 杂交增加了选择中性位点的等位基因数量; 引入具有选择优势的等位基因, 增加获得这些变异的类群的适合度, 促成快速的适应性转变(Choler et al, 2004; Martin et al, 2006; Castric et al, 2008; Kim et al, 2008). ...
Hybrid populations selectively filter gene introgression between species.
1
2001
... 越来越多的证据表明, 杂交普遍存在于生物类群分化和维持中, 具有重要的进化生物学意义.在农林业上, 杂交是一种重要的育种手段.通过人工杂交, 可以快速获得杂种优势、实现有益变异在不同育种材料间的重新组合, 甚至产生全新的表型.在自然类群中, 杂交也有类似的作用: 可以带来新的遗传变异, 且速度比突变要快得多(Anderson & Hubricht, 1938; Martinsen et al, 2001).因此, 杂交增加了选择中性位点的等位基因数量; 引入具有选择优势的等位基因, 增加获得这些变异的类群的适合度, 促成快速的适应性转变(Choler et al, 2004; Martin et al, 2006; Castric et al, 2008; Kim et al, 2008). ...
BaitFisher: a software package for multispecies target DNA enrichment probe design.
1
2016
... 尽管目标富集测序技术相对成熟, 但目前的应用较多地针对人类疾病研究, 在系统发育分析中的应用仍在发展.探针开发是一个重要环节, 从转录组或基因组开发探针的工具和流程已有不少报道(Mayer et al, 2016; Pavy et al, 2016; Schmickl et al, 2016).超保守序列(ultraconserved elements)也是开发捕获探针的重要来源, 目前哺乳动物(Bejerano et al, 2004; Reneker et al, 2012)、鸟类(Mccormack & Al, 2011)、两栖爬行类(Crawford et al, 2012)、昆虫(Branstetter et al, 2017)、高等植物(Freeling et al, 2009; Reneker et al, 2012)上的超保守序列已有报道.该技术还有一个特点, 就是通过对长目标序列多个不同区域设计探针, 可以实现对整条序列的捕获测序.这突破了目前流行的新一代测序的读长较短的限制.但这对数据分析也造成了问题, 因为目前流行的系统发育分析软件对大量长片段进行分析时计算速度往往很慢, 比如BEST (Edwards et al, 2007)和*BEST (Heled & Drummond, 2010). ...
Next-generation sequencing reveals phylogeographic structure and a species tree for recent bird divergences.
1
2011
... 尽管目标富集测序技术相对成熟, 但目前的应用较多地针对人类疾病研究, 在系统发育分析中的应用仍在发展.探针开发是一个重要环节, 从转录组或基因组开发探针的工具和流程已有不少报道(Mayer et al, 2016; Pavy et al, 2016; Schmickl et al, 2016).超保守序列(ultraconserved elements)也是开发捕获探针的重要来源, 目前哺乳动物(Bejerano et al, 2004; Reneker et al, 2012)、鸟类(Mccormack & Al, 2011)、两栖爬行类(Crawford et al, 2012)、昆虫(Branstetter et al, 2017)、高等植物(Freeling et al, 2009; Reneker et al, 2012)上的超保守序列已有报道.该技术还有一个特点, 就是通过对长目标序列多个不同区域设计探针, 可以实现对整条序列的捕获测序.这突破了目前流行的新一代测序的读长较短的限制.但这对数据分析也造成了问题, 因为目前流行的系统发育分析软件对大量长片段进行分析时计算速度往往很慢, 比如BEST (Edwards et al, 2007)和*BEST (Heled & Drummond, 2010). ...
A graphical method for detecting recombination in phylogenetic data sets.
1
1997
... 模型初选后, 应该用恰当的检验来评判选定模型的可靠性.这方面有一系列的工具可以利用, 比如位点内或位点间重组(Mcguire et al, 1997; Kosakovsky et al, 2006; Martin et al, 2015)、各种选择(Delport et al, 2010)、杂交(Yu et al, 2011)、时序性进化速率差异(heterotachy) (Pagel & Meade, 2008)和模型总体上的配合性(Albert & Schluter, 2005; Abby et al, 2012; Ackermann et al, 2016)等.如果初选模型不适合, 那么应该及时调整选用更恰当的模型.如果位点间基因树冲突, 那么可考虑选用应对杂交或者不完全谱系分离的模型, 而不是仍然将序列合并使用, 或者把不相配合的部分数据删除.当然还是要删除那些体现强烈选择印记的、变异饱和的(saturated) (Castresana, 2000; Rodríguezezpeleta et al, 2007; Gnirke et al, 2009)和包含有缺失值(missing)或有缺失变异(deletion)的位点(Lemmon et al, 2009). ...
Paralogs are revealed by proportion of heterozygotes and deviations in read ratios in genotyping-by-sequencing data from natural populations.
1
2016
... 相对传统技术来说, 这些分子标记技术的优势十分明显, 尽管数据处理中的某些具体环节还在发展, 但成熟的技术流程和相应的软件并不难获得.测序仪输出的原始数据转换、短读(short read)长数据质控、短读长数据的比对、转录组或基因组的组装、单碱基突变或单倍型获取、各步骤质量评估和数据筛选等重要环节的分析对一般的系统发育和群体遗传学的研究团队并不难实现, 有大量的高质量技术指南可以参考(DePristo et al, 2011; Nielsen et al, 2011).对某一种特定分子标记技术的分析策略也有不少参考, 比如RAD测序方面(Hapke & Thiele, 2016; Kim et al, 2016; McKinney et al, 2016; Shafer et al, 2016; Torkamaneh et al, 2016, 2017). ...
A genealogical interpretation of principal components analysis.
Ancient hybridization fuels rapid cichlid fish adaptive radiations.
1
2017
... F4-ratio检验可以用来推断杂交造成的遗传重组的比例, 即使在不知道祖先群体的情况下, 也可以依据对系统发育关系的假定进行推断.四群体检验不但可以对杂交是否存在给出证据, 还可以提供基因流的方向.这些四群体检验也同样被广泛用于检测现代人类基因组中的古人类成分和相关的历史过程(Green et al, 2010; Fu et al, 2014, 2016; Prufer et al, 2014; Meyer et al, 2016).最近对非洲丽鱼的研究将该方法进一步拓展为五群体检验, 或称为f5-statistic (Meier et al, 2017), 该研究表明古老的杂交事件带来遗传变异, 成为后期适应性分化和物种形成的重要推动力. ...
Genotype calling and phasing using next- generation sequencing reads and a haplotype scaffold.
Synthetic maps of human gene frequencies in Europeans.
1
1978
... 主成分分析(principal component analysis, PCA)是一类经典的多元统计技术, 很早就开始被应用在遗传数据分析中(Menozzi et al, 1978).主成分分析可以将样本间的关系呈现在主成分形成的二维或三维空间上, 但它不能提供杂交存在与否的检验(Patterson et al, 2012), 介于类群间的样本不一定就来自于类群间的杂交(Yang et al, 2012).因此, EIGENSTRAT等软件基于PCA来估算杂交(Price et al, 2006)可能会得出错误的结果.除了EIGENSTRAT, adegenet也是一个主成分分析的工具(Jombart, 2008). ...
A high-coverage genome sequence from an archaic Denisovan individual.
1
2012
... 重组图(admixture graph)是一个允许类群间基因交流的系统发育树的拓展.基于上面提到的F-statistics就可以构建这样的图, 目前有几种工具可以利用.MixMapper (Lipson et al, 2014)是一个半自动的工具.它首先基于由一对明显没有杂交的群体间等位基因频率(F2)求算的遗传聚类生成邻接树(neighbor-joining tree), 然后在允许基因流的情况下把剩下的群体添加到图上来(Lipson et al, 2013, 2014).TreeMix (Pickrell & Pritchard, 2012)实现了与MixMapper在理论上类似的算法.在假定重组或者基因流个数的情况下, 它可以全自动地计算出重组图.不同的重组次数的假定会对结果带来极大影响, 选取时需谨慎.上面提到的ADMIXTOOLS软件(Patterson et al, 2012)中也提供了类似的工具.但这里的重组图工具更加稳健, 因为它提供了假设中的重组模式是否和数据相符的检验.重组图的手段已经在人类遗传领域广泛应用, 曾被用来揭示北美原著民的遗传组成和来源(Raghavan et al, 2014b)、丹尼索瓦人(Denisovans)对现代人群的遗传贡献(Meyer et al, 2012)、新世界寒带地区的人类定居历史(Raghavan et al, 2014a). ...
Nuclear DNA sequences from the Middle Pleistocene Sima de los Huesos hominins.
1
2016
... F4-ratio检验可以用来推断杂交造成的遗传重组的比例, 即使在不知道祖先群体的情况下, 也可以依据对系统发育关系的假定进行推断.四群体检验不但可以对杂交是否存在给出证据, 还可以提供基因流的方向.这些四群体检验也同样被广泛用于检测现代人类基因组中的古人类成分和相关的历史过程(Green et al, 2010; Fu et al, 2014, 2016; Prufer et al, 2014; Meyer et al, 2016).最近对非洲丽鱼的研究将该方法进一步拓展为五群体检验, 或称为f5-statistic (Meier et al, 2017), 该研究表明古老的杂交事件带来遗传变异, 成为后期适应性分化和物种形成的重要推动力. ...
The history of African gene flow into southern Europeans, Levantines, and Jews.
1
2011
... 上面提到的主成分分析、聚类分析(比如基于structure和admixture的分析)可以用来澄清群体遗传结构, 并对杂交的存在提供全局性的线索(那些看似杂交的迹象可能由杂交以外的其他因素导致), 但它们都没有提供杂交是否存在的检验.比如, 与距离关联的分化(isolation by distance)可以产生在PCA上的梯度变异.基于structure和admixture的结果也很难做出对群体历史的推算, 因为它们没有对特定的群体历史模型进行检验, 而是简单地假定抽样群体都是从某特定群体快速辐射分化而来.Patterson等(2012)综合已有工作(Reich et al, 2009; Green et al, 2010; Durand et al, 2011; Moorjani et al, 2011), 归纳出了几个针对群体间杂交历史的检验和相应的参数, 并提供了实现有关计算的软件包(ADMIXTOOLS).这些参数和检验包括1个三群体检验(f3-statistic, the three-population test)和2个四群体检验(D-statistics或者ABBA-BABA test, 以及f4- statistics或F4-ratio).这里的f3-statistic和f4-statistics也被统称为F-statistics. ...
Reconstructing the population genetic history of the Caribbean.
1
2013
... 已经有一些估算位点祖先状态的计算工具, Padhukasahasram (2014)就进行了较为详尽的综述.一般来说, 这些计算工具需要借助隐马尔科夫模型(hidden Markov models, HMMs)来确定不同亲本来源的基因组区段的界限.HAPMIX软件可以利用未定相(unphased)的基因型数据确定杂种个体特定位点的祖先来源(Price et al, 2009).CRFs应用一个广义隐马尔科夫模型(generalized hidden Markov model), 借助训练集数据实现了对祖先来源的估算(Sankararaman et al, 2012).实现这类分析的软件还包括: SABER (Tang et al, 2006)、HAPA (Sundquist et al, 2008)、LAMP (Sankararaman et al, 2008)、LAMP- LD/LAMP-HAP (Baran et al, 2012)、WINPOP (Pasaniuc et al, 2009)、SupportMix (Omberg et al, 2012)、ASPCA (Moreno-Estrada et al, 2013)、ALLOY (Rodriguez et al, 2013)、RFMix (Maples et al, 2013)、Lanc- CSV (Brown & Pasaniuc, 2014)和EILA (Yang et al, 2013).EILA没有对输入数据位点间连锁平衡的前提假设, 且比LAMP和HAPMIX更准确, 运行速度也不慢.Lanc-CSV实现了经典算法, 但对超大样本数据有速度上的优势. ...
Detection of interspecies hybridisation in Chondrichthyes: hybrids and hybrid offspring between Australian (Carcharhinus tilstoni) and common (C. limbatus) blacktip shark found in an Australian fishery.
1
2012
... 同时, 对只关注少量位点但包含多个个体的项目, 扩增子测序有很大的吸引力(Griffin et al, 2011).在检测杂交存在方面, 一个典型应用实例是鲨鱼的研究.研究人员通过扩增子测序建立的分子标记技术, 检测到帝氏真鲨(Carcharchinus tilstoni)和黑边鳍真鲨(C. limbatus)间的杂交(Morgan et al, 2012).目前, 这项技术策略还被用于获取来自全线粒体基因组(Chan et al, 2010; Morin et al, 2010; Gunnarsdóttir et al, 2011)、全叶绿体基因组(Parks, 2009)、宏基因组(metagenomics)遗传变异的研究中. ...
... 同时, 对只关注少量位点但包含多个个体的项目, 扩增子测序有很大的吸引力(Griffin et al, 2011).在检测杂交存在方面, 一个典型应用实例是鲨鱼的研究.研究人员通过扩增子测序建立的分子标记技术, 检测到帝氏真鲨(Carcharchinus tilstoni)和黑边鳍真鲨(C. limbatus)间的杂交(Morgan et al, 2012).目前, 这项技术策略还被用于获取来自全线粒体基因组(Chan et al, 2010; Morin et al, 2010; Gunnarsdóttir et al, 2011)、全叶绿体基因组(Parks, 2009)、宏基因组(metagenomics)遗传变异的研究中. ...
... 转录组测序(transcriptome sequencing)或者RNA测序(RNA sequencing)是针对基因组中的表达序列进行测序, 本质上也是一种简化基因组测序技术(Marioni et al, 2008; Morin et al, 2008; Wang et al, 2009).尽管这项技术通常被用于适应性进化和生态基因组学研究, 但已有的研究实例表明, 该技术获得的转录组序列可以用于重建系统发育和检测杂交的存在(Nabholz et al, 2011; Pease et al, 2016).此外, 该技术对发掘单碱基突变、进行群体遗传研究也很有吸引力, 因为它不仅可实现对复杂基因组的简化, 而且提供了来自直接与功能相关转录序列的信息.从转录组组装序列中寻找直系同源基因的计算工具已有报道, HaMStR (Ebersberger et al, 2009)是比较不错的一个.该分子标记技术的特征参见图1. ...
Estimating species trees: practical and theoretical aspects.
Dynamic evolution of base composition: causes and consequences in avian phylogenomics.
1
2011
... 转录组测序(transcriptome sequencing)或者RNA测序(RNA sequencing)是针对基因组中的表达序列进行测序, 本质上也是一种简化基因组测序技术(Marioni et al, 2008; Morin et al, 2008; Wang et al, 2009).尽管这项技术通常被用于适应性进化和生态基因组学研究, 但已有的研究实例表明, 该技术获得的转录组序列可以用于重建系统发育和检测杂交的存在(Nabholz et al, 2011; Pease et al, 2016).此外, 该技术对发掘单碱基突变、进行群体遗传研究也很有吸引力, 因为它不仅可实现对复杂基因组的简化, 而且提供了来自直接与功能相关转录序列的信息.从转录组组装序列中寻找直系同源基因的计算工具已有报道, HaMStR (Ebersberger et al, 2009)是比较不错的一个.该分子标记技术的特征参见图1. ...
Computational approaches to species phylogeny inference and gene tree reconciliation.
... 生物类群中杂交的普遍存在获得了越来越多的认可.然而, 相对于复杂的生物多样性, 清晰描述的杂交事件仍显得非常少.实际上, 作为一个重要的进化过程和生物多样性维持机制, 杂交受到的关注还远远不够.据估算, 有25%的高等植物物种(Mallet, 2005)和10%的动物物种(Schwenk et al, 2008)间存在着种间杂交; 此外, 在真菌(Nelson, 1963; Depotter et al, 2016)、细菌(Duncan et al, 1989; Cohan & Kane, 2001; Cohan, 2002; Earl et al, 2008)、病毒(Worobey & Holmes, 1999; Bujarski, 2013; Lefeuvre & Moriones, 2015; Pérez-Losada et al, 2015; Su et al, 2016)的基因组进化中, 杂交普遍存在.以植物为例, Flora of China共记载我国3万余种植物(Wu et al, 2014), 按25%计算, 我们推测其中9,000种植物间存在杂交.同时, 日益加剧的全球气候变化和人类活动将对生物多样性的分布特征产生显著影响, 进而改变种间关系, 导致原已形成的种间生殖隔离改变或降低, 进一步增加杂交的机会(Stenz et al, 2015; Novikova et al, 2016; Vallejo- Marin & Hiscock, 2016).相较于上面的估算, 得到确认的植物杂交案例非常稀少, 我们认为可能不足1%.因此, 要全面澄清杂交产生的机制、杂交对多样性形成和维持的影响及其进化生物学意义还为时尚早.目前已经开展或正在进行的工作所覆盖的杂交事件, 估计不超过总量的1‰.检测杂交及评估其发生的范围和机制, 将成为未来生物多样性监测、管理和保护中不可缺少的组成部分.在后志书时代的今天, 检测杂交等遗传交换事件的存在, 进而揭示它对生物多样性形成和维持的重要价值, 是我们面临的重要挑战. ...
Genotype and SNP calling from next-generation sequencing data.
1
2011
... 相对传统技术来说, 这些分子标记技术的优势十分明显, 尽管数据处理中的某些具体环节还在发展, 但成熟的技术流程和相应的软件并不难获得.测序仪输出的原始数据转换、短读(short read)长数据质控、短读长数据的比对、转录组或基因组的组装、单碱基突变或单倍型获取、各步骤质量评估和数据筛选等重要环节的分析对一般的系统发育和群体遗传学的研究团队并不难实现, 有大量的高质量技术指南可以参考(DePristo et al, 2011; Nielsen et al, 2011).对某一种特定分子标记技术的分析策略也有不少参考, 比如RAD测序方面(Hapke & Thiele, 2016; Kim et al, 2016; McKinney et al, 2016; Shafer et al, 2016; Torkamaneh et al, 2016, 2017). ...
The genes underlying the process of speciation.
1
2011
... 与杂交关联的是不同特征的基因组间的重组, 这种重组会带来一个嵌合的基因组, 在嵌合的基因组上不同的区域体现着不同的进化历程.识别这些位点不但对澄清种间或群体间的基因交流有意义(Green et al, 2010; Reich et al, 2010; Prufer et al, 2014), 同时也可以提供重组模式和适应性分化的信息(Kim & Rothschild, 2014).这些位点的识别对于揭示生殖隔离的遗传基础和找寻物种形成基因(speciation genes)有重要意义(Wu & Ting, 2004; Nosil & Schluter, 2011; Burri et al, 2015), 同时对建立针对濒危物种的保护策略有应用价值(der Sarkissian et al, 2015).基于基因组水平的滑动窗(sliding window)分析可以追溯不同染色体区段的祖先状态, 上面提到过的D-statistics就是一个很好的工具(Kronforst et al, 2013; Smith & Kronforst, 2013). ...
Sequencing of the genus Arabidopsis identifies a complex history of nonbifurcating speciation and abundant trans-specific polymorphism.
2
2016
... 生物类群中杂交的普遍存在获得了越来越多的认可.然而, 相对于复杂的生物多样性, 清晰描述的杂交事件仍显得非常少.实际上, 作为一个重要的进化过程和生物多样性维持机制, 杂交受到的关注还远远不够.据估算, 有25%的高等植物物种(Mallet, 2005)和10%的动物物种(Schwenk et al, 2008)间存在着种间杂交; 此外, 在真菌(Nelson, 1963; Depotter et al, 2016)、细菌(Duncan et al, 1989; Cohan & Kane, 2001; Cohan, 2002; Earl et al, 2008)、病毒(Worobey & Holmes, 1999; Bujarski, 2013; Lefeuvre & Moriones, 2015; Pérez-Losada et al, 2015; Su et al, 2016)的基因组进化中, 杂交普遍存在.以植物为例, Flora of China共记载我国3万余种植物(Wu et al, 2014), 按25%计算, 我们推测其中9,000种植物间存在杂交.同时, 日益加剧的全球气候变化和人类活动将对生物多样性的分布特征产生显著影响, 进而改变种间关系, 导致原已形成的种间生殖隔离改变或降低, 进一步增加杂交的机会(Stenz et al, 2015; Novikova et al, 2016; Vallejo- Marin & Hiscock, 2016).相较于上面的估算, 得到确认的植物杂交案例非常稀少, 我们认为可能不足1%.因此, 要全面澄清杂交产生的机制、杂交对多样性形成和维持的影响及其进化生物学意义还为时尚早.目前已经开展或正在进行的工作所覆盖的杂交事件, 估计不超过总量的1‰.检测杂交及评估其发生的范围和机制, 将成为未来生物多样性监测、管理和保护中不可缺少的组成部分.在后志书时代的今天, 检测杂交等遗传交换事件的存在, 进而揭示它对生物多样性形成和维持的重要价值, 是我们面临的重要挑战. ...
... 这个技术的实现是显而易见的.对于已有参考基因组、基因组大小合适的类群, 全基因组测序(更多的是基因组重测序, genome resequencing)应用已经很常见了, 尤其是对近缘种间或种内群体分化的研究中, 其优势更加明显.人类的1,000 Genomes计划(http://www.internationalgenome.org/)(The Genomes Project Consortium et al, 2010)和植物中拟南芥(Arabidopsis thaliana)的1,001 Genomes计划(http://1001genomes.org/) (Weigel & Mott, 2009)都为我们作了很好的示范.值得一提的是, 基因组水平的证据表明种间遗传交换参与了人(Patterson et al, 2012; Hellenthal et al, 2014; Lazaridis et al, 2014; Sankararaman et al, 2014; Ackermann et al, 2016)和拟南芥(Stenz et al, 2015; Novikova et al, 2016)的起源和分化. ...
Inferring genome-wide patterns of admixture in Qataris using fifty-five ancestral populations.
1
2012
... 已经有一些估算位点祖先状态的计算工具, Padhukasahasram (2014)就进行了较为详尽的综述.一般来说, 这些计算工具需要借助隐马尔科夫模型(hidden Markov models, HMMs)来确定不同亲本来源的基因组区段的界限.HAPMIX软件可以利用未定相(unphased)的基因型数据确定杂种个体特定位点的祖先来源(Price et al, 2009).CRFs应用一个广义隐马尔科夫模型(generalized hidden Markov model), 借助训练集数据实现了对祖先来源的估算(Sankararaman et al, 2012).实现这类分析的软件还包括: SABER (Tang et al, 2006)、HAPA (Sundquist et al, 2008)、LAMP (Sankararaman et al, 2008)、LAMP- LD/LAMP-HAP (Baran et al, 2012)、WINPOP (Pasaniuc et al, 2009)、SupportMix (Omberg et al, 2012)、ASPCA (Moreno-Estrada et al, 2013)、ALLOY (Rodriguez et al, 2013)、RFMix (Maples et al, 2013)、Lanc- CSV (Brown & Pasaniuc, 2014)和EILA (Yang et al, 2013).EILA没有对输入数据位点间连锁平衡的前提假设, 且比LAMP和HAPMIX更准确, 运行速度也不慢.Lanc-CSV实现了经典算法, 但对超大样本数据有速度上的优势. ...
Parallel tagged amplicon sequencing reveals major lineages and phylogenetic structure in the North American tiger salamander (Ambystoma tigrinum) species complex.
1
2013
... 基因组技术带来了海量数据, 同时也伴随着一些问题.有专家认为, 随着谱系基因组学的数据量增大, 潜在的误差来源也会增加(Philippe et al, 2005).误差来源主要有两个: 随机误差(stochastic error)和系统误差(systematic error) (Swofford et al, 1996).随机误差是由数据信息量不足造成的, 应该会随着数据量的增加而降低; 系统误差主要来自于模型的选配, 会随着数据量的增加而增大(Philippe et al, 2005; Kumar, 2012).如果没能很好地控制系统误差, 我们可能得到看似支持率很好但错误的系统发育重建(O’Neill et al, 2013).当仅利用一个基因位点构建基因树时, 增加序列长度可以降低随机误差; 但如果没有考虑增加序列长度带来的位点内重组的风险, 那么会相应地增加系统误差.当试图把不同位点合并成一个位点进而构建基因树时, 同样的问题也会发生. ...
RNA sequencing: advances, challenges and opportunities.
Modelling heterotachy in phylogenetic inference by reversible-jump Markov chain Monte Carlo.
1
2008
... 模型初选后, 应该用恰当的检验来评判选定模型的可靠性.这方面有一系列的工具可以利用, 比如位点内或位点间重组(Mcguire et al, 1997; Kosakovsky et al, 2006; Martin et al, 2015)、各种选择(Delport et al, 2010)、杂交(Yu et al, 2011)、时序性进化速率差异(heterotachy) (Pagel & Meade, 2008)和模型总体上的配合性(Albert & Schluter, 2005; Abby et al, 2012; Ackermann et al, 2016)等.如果初选模型不适合, 那么应该及时调整选用更恰当的模型.如果位点间基因树冲突, 那么可考虑选用应对杂交或者不完全谱系分离的模型, 而不是仍然将序列合并使用, 或者把不相配合的部分数据删除.当然还是要删除那些体现强烈选择印记的、变异饱和的(saturated) (Castresana, 2000; Rodríguezezpeleta et al, 2007; Gnirke et al, 2009)和包含有缺失值(missing)或有缺失变异(deletion)的位点(Lemmon et al, 2009). ...
Increasing phylogenetic resolution at low taxonomic levels using massively parallel sequencing of chloroplast genomes.
1
2009
... 同时, 对只关注少量位点但包含多个个体的项目, 扩增子测序有很大的吸引力(Griffin et al, 2011).在检测杂交存在方面, 一个典型应用实例是鲨鱼的研究.研究人员通过扩增子测序建立的分子标记技术, 检测到帝氏真鲨(Carcharchinus tilstoni)和黑边鳍真鲨(C. limbatus)间的杂交(Morgan et al, 2012).目前, 这项技术策略还被用于获取来自全线粒体基因组(Chan et al, 2010; Morin et al, 2010; Gunnarsdóttir et al, 2011)、全叶绿体基因组(Parks, 2009)、宏基因组(metagenomics)遗传变异的研究中. ...
Inference of locus-specific ancestry in closely related populations.
1
2009
... 已经有一些估算位点祖先状态的计算工具, Padhukasahasram (2014)就进行了较为详尽的综述.一般来说, 这些计算工具需要借助隐马尔科夫模型(hidden Markov models, HMMs)来确定不同亲本来源的基因组区段的界限.HAPMIX软件可以利用未定相(unphased)的基因型数据确定杂种个体特定位点的祖先来源(Price et al, 2009).CRFs应用一个广义隐马尔科夫模型(generalized hidden Markov model), 借助训练集数据实现了对祖先来源的估算(Sankararaman et al, 2012).实现这类分析的软件还包括: SABER (Tang et al, 2006)、HAPA (Sundquist et al, 2008)、LAMP (Sankararaman et al, 2008)、LAMP- LD/LAMP-HAP (Baran et al, 2012)、WINPOP (Pasaniuc et al, 2009)、SupportMix (Omberg et al, 2012)、ASPCA (Moreno-Estrada et al, 2013)、ALLOY (Rodriguez et al, 2013)、RFMix (Maples et al, 2013)、Lanc- CSV (Brown & Pasaniuc, 2014)和EILA (Yang et al, 2013).EILA没有对输入数据位点间连锁平衡的前提假设, 且比LAMP和HAPMIX更准确, 运行速度也不慢.Lanc-CSV实现了经典算法, 但对超大样本数据有速度上的优势. ...
Ancient admixture in human history.
4
2012
... 这个技术的实现是显而易见的.对于已有参考基因组、基因组大小合适的类群, 全基因组测序(更多的是基因组重测序, genome resequencing)应用已经很常见了, 尤其是对近缘种间或种内群体分化的研究中, 其优势更加明显.人类的1,000 Genomes计划(http://www.internationalgenome.org/)(The Genomes Project Consortium et al, 2010)和植物中拟南芥(Arabidopsis thaliana)的1,001 Genomes计划(http://1001genomes.org/) (Weigel & Mott, 2009)都为我们作了很好的示范.值得一提的是, 基因组水平的证据表明种间遗传交换参与了人(Patterson et al, 2012; Hellenthal et al, 2014; Lazaridis et al, 2014; Sankararaman et al, 2014; Ackermann et al, 2016)和拟南芥(Stenz et al, 2015; Novikova et al, 2016)的起源和分化. ...
... 主成分分析(principal component analysis, PCA)是一类经典的多元统计技术, 很早就开始被应用在遗传数据分析中(Menozzi et al, 1978).主成分分析可以将样本间的关系呈现在主成分形成的二维或三维空间上, 但它不能提供杂交存在与否的检验(Patterson et al, 2012), 介于类群间的样本不一定就来自于类群间的杂交(Yang et al, 2012).因此, EIGENSTRAT等软件基于PCA来估算杂交(Price et al, 2006)可能会得出错误的结果.除了EIGENSTRAT, adegenet也是一个主成分分析的工具(Jombart, 2008). ...
... 重组图(admixture graph)是一个允许类群间基因交流的系统发育树的拓展.基于上面提到的F-statistics就可以构建这样的图, 目前有几种工具可以利用.MixMapper (Lipson et al, 2014)是一个半自动的工具.它首先基于由一对明显没有杂交的群体间等位基因频率(F2)求算的遗传聚类生成邻接树(neighbor-joining tree), 然后在允许基因流的情况下把剩下的群体添加到图上来(Lipson et al, 2013, 2014).TreeMix (Pickrell & Pritchard, 2012)实现了与MixMapper在理论上类似的算法.在假定重组或者基因流个数的情况下, 它可以全自动地计算出重组图.不同的重组次数的假定会对结果带来极大影响, 选取时需谨慎.上面提到的ADMIXTOOLS软件(Patterson et al, 2012)中也提供了类似的工具.但这里的重组图工具更加稳健, 因为它提供了假设中的重组模式是否和数据相符的检验.重组图的手段已经在人类遗传领域广泛应用, 曾被用来揭示北美原著民的遗传组成和来源(Raghavan et al, 2014b)、丹尼索瓦人(Denisovans)对现代人群的遗传贡献(Meyer et al, 2012)、新世界寒带地区的人类定居历史(Raghavan et al, 2014a). ...
Development of highly reliable in silico SNP resource and genotyping assay from exome capture and sequencing: an example from black spruce (Picea mariana).
1
2016
... 尽管目标富集测序技术相对成熟, 但目前的应用较多地针对人类疾病研究, 在系统发育分析中的应用仍在发展.探针开发是一个重要环节, 从转录组或基因组开发探针的工具和流程已有不少报道(Mayer et al, 2016; Pavy et al, 2016; Schmickl et al, 2016).超保守序列(ultraconserved elements)也是开发捕获探针的重要来源, 目前哺乳动物(Bejerano et al, 2004; Reneker et al, 2012)、鸟类(Mccormack & Al, 2011)、两栖爬行类(Crawford et al, 2012)、昆虫(Branstetter et al, 2017)、高等植物(Freeling et al, 2009; Reneker et al, 2012)上的超保守序列已有报道.该技术还有一个特点, 就是通过对长目标序列多个不同区域设计探针, 可以实现对整条序列的捕获测序.这突破了目前流行的新一代测序的读长较短的限制.但这对数据分析也造成了问题, 因为目前流行的系统发育分析软件对大量长片段进行分析时计算速度往往很慢, 比如BEST (Edwards et al, 2007)和*BEST (Heled & Drummond, 2010). ...
A genomic perspective on hybridization and speciation.
Phylogenomics reveals three sources of adaptive variation during a rapid radiation.
1
2016
... 转录组测序(transcriptome sequencing)或者RNA测序(RNA sequencing)是针对基因组中的表达序列进行测序, 本质上也是一种简化基因组测序技术(Marioni et al, 2008; Morin et al, 2008; Wang et al, 2009).尽管这项技术通常被用于适应性进化和生态基因组学研究, 但已有的研究实例表明, 该技术获得的转录组序列可以用于重建系统发育和检测杂交的存在(Nabholz et al, 2011; Pease et al, 2016).此外, 该技术对发掘单碱基突变、进行群体遗传研究也很有吸引力, 因为它不仅可实现对复杂基因组的简化, 而且提供了来自直接与功能相关转录序列的信息.从转录组组装序列中寻找直系同源基因的计算工具已有报道, HaMStR (Ebersberger et al, 2009)是比较不错的一个.该分子标记技术的特征参见图1. ...
Double digest RADseq: an inexpensive method for de novo SNP discovery and genotyping in model and non-model species.
1
2012
... 这类技术都包含一个重要步骤, 即对目标样本基因组进行限制性内切酶酶切, 有些还包含对酶切产物片段大小的筛选(Baird et al, 2008; van Tassell et al, 2008; Kim et al, 2016).酶切过程中, 可用单一的内切酶, 也可用两个内切酶的组合(Peterson et al, 2012); 酶切的特征可能是甲基化敏感的、甲基化不敏感的或IIB型限制性核酸内切酶(Wang et al, 2012).酶的组合和酶切片段筛选给了这类技术极大的灵活性, 加上样本标签后可以实现各种组合的混样测序, 这类技术的潜力巨大.基于类似的简化原理, 已有大量特定技术衍生出来(如reduced-representation library sequencing (RRL), restriction-site- associated DNA sequencing (RAD), genotyping by sequencing (GBS)), 有不少综述性论文对它们进行了详细介绍(Davey et al, 2011; Andrews et al, 2016).该分子标记技术的特征参见图1. ...
Recombination in viruses: mechanisms, methods of study, and evolutionary consequences
1
2015
... 生物类群中杂交的普遍存在获得了越来越多的认可.然而, 相对于复杂的生物多样性, 清晰描述的杂交事件仍显得非常少.实际上, 作为一个重要的进化过程和生物多样性维持机制, 杂交受到的关注还远远不够.据估算, 有25%的高等植物物种(Mallet, 2005)和10%的动物物种(Schwenk et al, 2008)间存在着种间杂交; 此外, 在真菌(Nelson, 1963; Depotter et al, 2016)、细菌(Duncan et al, 1989; Cohan & Kane, 2001; Cohan, 2002; Earl et al, 2008)、病毒(Worobey & Holmes, 1999; Bujarski, 2013; Lefeuvre & Moriones, 2015; Pérez-Losada et al, 2015; Su et al, 2016)的基因组进化中, 杂交普遍存在.以植物为例, Flora of China共记载我国3万余种植物(Wu et al, 2014), 按25%计算, 我们推测其中9,000种植物间存在杂交.同时, 日益加剧的全球气候变化和人类活动将对生物多样性的分布特征产生显著影响, 进而改变种间关系, 导致原已形成的种间生殖隔离改变或降低, 进一步增加杂交的机会(Stenz et al, 2015; Novikova et al, 2016; Vallejo- Marin & Hiscock, 2016).相较于上面的估算, 得到确认的植物杂交案例非常稀少, 我们认为可能不足1%.因此, 要全面澄清杂交产生的机制、杂交对多样性形成和维持的影响及其进化生物学意义还为时尚早.目前已经开展或正在进行的工作所覆盖的杂交事件, 估计不超过总量的1‰.检测杂交及评估其发生的范围和机制, 将成为未来生物多样性监测、管理和保护中不可缺少的组成部分.在后志书时代的今天, 检测杂交等遗传交换事件的存在, 进而揭示它对生物多样性形成和维持的重要价值, 是我们面临的重要挑战. ...
Phylogenomics
2
2005
... 基因组技术带来了海量数据, 同时也伴随着一些问题.有专家认为, 随着谱系基因组学的数据量增大, 潜在的误差来源也会增加(Philippe et al, 2005).误差来源主要有两个: 随机误差(stochastic error)和系统误差(systematic error) (Swofford et al, 1996).随机误差是由数据信息量不足造成的, 应该会随着数据量的增加而降低; 系统误差主要来自于模型的选配, 会随着数据量的增加而增大(Philippe et al, 2005; Kumar, 2012).如果没能很好地控制系统误差, 我们可能得到看似支持率很好但错误的系统发育重建(O’Neill et al, 2013).当仅利用一个基因位点构建基因树时, 增加序列长度可以降低随机误差; 但如果没有考虑增加序列长度带来的位点内重组的风险, 那么会相应地增加系统误差.当试图把不同位点合并成一个位点进而构建基因树时, 同样的问题也会发生. ...
... ).随机误差是由数据信息量不足造成的, 应该会随着数据量的增加而降低; 系统误差主要来自于模型的选配, 会随着数据量的增加而增大(Philippe et al, 2005; Kumar, 2012).如果没能很好地控制系统误差, 我们可能得到看似支持率很好但错误的系统发育重建(O’Neill et al, 2013).当仅利用一个基因位点构建基因树时, 增加序列长度可以降低随机误差; 但如果没有考虑增加序列长度带来的位点内重组的风险, 那么会相应地增加系统误差.当试图把不同位点合并成一个位点进而构建基因树时, 同样的问题也会发生. ...
Inference of population splits and mixtures from genome-wide allele frequency data.
1
2012
... 重组图(admixture graph)是一个允许类群间基因交流的系统发育树的拓展.基于上面提到的F-statistics就可以构建这样的图, 目前有几种工具可以利用.MixMapper (Lipson et al, 2014)是一个半自动的工具.它首先基于由一对明显没有杂交的群体间等位基因频率(F2)求算的遗传聚类生成邻接树(neighbor-joining tree), 然后在允许基因流的情况下把剩下的群体添加到图上来(Lipson et al, 2013, 2014).TreeMix (Pickrell & Pritchard, 2012)实现了与MixMapper在理论上类似的算法.在假定重组或者基因流个数的情况下, 它可以全自动地计算出重组图.不同的重组次数的假定会对结果带来极大影响, 选取时需谨慎.上面提到的ADMIXTOOLS软件(Patterson et al, 2012)中也提供了类似的工具.但这里的重组图工具更加稳健, 因为它提供了假设中的重组模式是否和数据相符的检验.重组图的手段已经在人类遗传领域广泛应用, 曾被用来揭示北美原著民的遗传组成和来源(Raghavan et al, 2014b)、丹尼索瓦人(Denisovans)对现代人群的遗传贡献(Meyer et al, 2012)、新世界寒带地区的人类定居历史(Raghavan et al, 2014a). ...
Principal components analysis corrects for stratification in genome-wide association studies.
1
2006
... 主成分分析(principal component analysis, PCA)是一类经典的多元统计技术, 很早就开始被应用在遗传数据分析中(Menozzi et al, 1978).主成分分析可以将样本间的关系呈现在主成分形成的二维或三维空间上, 但它不能提供杂交存在与否的检验(Patterson et al, 2012), 介于类群间的样本不一定就来自于类群间的杂交(Yang et al, 2012).因此, EIGENSTRAT等软件基于PCA来估算杂交(Price et al, 2006)可能会得出错误的结果.除了EIGENSTRAT, adegenet也是一个主成分分析的工具(Jombart, 2008). ...
Sensitive detection of chromosomal segments of distinct ancestry in admixed populations.
1
2009
... 已经有一些估算位点祖先状态的计算工具, Padhukasahasram (2014)就进行了较为详尽的综述.一般来说, 这些计算工具需要借助隐马尔科夫模型(hidden Markov models, HMMs)来确定不同亲本来源的基因组区段的界限.HAPMIX软件可以利用未定相(unphased)的基因型数据确定杂种个体特定位点的祖先来源(Price et al, 2009).CRFs应用一个广义隐马尔科夫模型(generalized hidden Markov model), 借助训练集数据实现了对祖先来源的估算(Sankararaman et al, 2012).实现这类分析的软件还包括: SABER (Tang et al, 2006)、HAPA (Sundquist et al, 2008)、LAMP (Sankararaman et al, 2008)、LAMP- LD/LAMP-HAP (Baran et al, 2012)、WINPOP (Pasaniuc et al, 2009)、SupportMix (Omberg et al, 2012)、ASPCA (Moreno-Estrada et al, 2013)、ALLOY (Rodriguez et al, 2013)、RFMix (Maples et al, 2013)、Lanc- CSV (Brown & Pasaniuc, 2014)和EILA (Yang et al, 2013).EILA没有对输入数据位点间连锁平衡的前提假设, 且比LAMP和HAPMIX更准确, 运行速度也不慢.Lanc-CSV实现了经典算法, 但对超大样本数据有速度上的优势. ...
Inference of population structure using multilocus genotype data.
1
2000
... 这里提到的聚类方法(clustering methods)是指按等位基因频率分布模式, 将个体的来源归入相应祖先群体(ancestral populations)中.简单地说, 聚类的基本过程是: 先假定有K个可能存在的祖先群体; 然后依据个体基因型计算它们被判别为各群体的概率; 在计算时, 假定各位点群体上的频率分布服从哈代-温伯格平衡(Hardy-Weinberg equilibrium).利用structure (Pritchard et al, 2000; Falush et al, 2003)和admixture (Alexander et al, 2009)等基于模型的聚类方法对每一个参试个体估算其来自于K个祖先群体中某个特定群体的遗传比例.祖先群体的数量K是个先验值, 可以依据一些已知的研究背景确定, 也可以通过最小K值的策略估算出来.旨在揭示杂交的群体遗传研究中, 此类聚类方法的应用非常普遍.InStruct是structure软件的拓展, 它没有哈代-温伯格平衡的前提假设, 可以同时对群体结构和近交率进行估算(Gao et al, 2007), 这对近交或自交比较频繁的植物研究有利用价值.类似的聚类运算的软件还有不少, 例如FRAPPE (Tang et al, 2007)、sNMF (Frichot et al, 2014)、Geneland (Guillot et al, 2004, 2005)等.它们在操作上类似, 运算速度相差不少, 有些可以考虑样本群体间的环境或地理信息.这类聚类方法可以应用于基因型数据、二倍化的单倍型数据(pseudo-homozygous genotype)或者基因型的似然值数据(genotype likelihood).ANGSD软件包中的NGSAdmix软件(Skotte et al, 2013)可以基于基因组水平基因型的似然值数据而不是基因型数据做聚类分析. ...
The complete genome sequence of a Neanderthal from the Altai Mountains.
3
2014
... 三群体检验基于对群体间等位基因频率关联性进行评估, 可以对群体间即便是非常近期发生的杂交事件给出清晰的验证.研究人员成功地利用f3-statistic估算出尼安德特人(Homo neanderthalensis)贡献了非洲现代人群1.5-2.1%的遗传变异(Prufer et al, 2014).通过选用外类群, f3-statistic也被用来揭示多个不同类群群体的多重起源历史(Haak et al, 2015; Haber et al, 2016; Mörseburg et al, 2016). ...
... F4-ratio检验可以用来推断杂交造成的遗传重组的比例, 即使在不知道祖先群体的情况下, 也可以依据对系统发育关系的假定进行推断.四群体检验不但可以对杂交是否存在给出证据, 还可以提供基因流的方向.这些四群体检验也同样被广泛用于检测现代人类基因组中的古人类成分和相关的历史过程(Green et al, 2010; Fu et al, 2014, 2016; Prufer et al, 2014; Meyer et al, 2016).最近对非洲丽鱼的研究将该方法进一步拓展为五群体检验, 或称为f5-statistic (Meier et al, 2017), 该研究表明古老的杂交事件带来遗传变异, 成为后期适应性分化和物种形成的重要推动力. ...
... 与杂交关联的是不同特征的基因组间的重组, 这种重组会带来一个嵌合的基因组, 在嵌合的基因组上不同的区域体现着不同的进化历程.识别这些位点不但对澄清种间或群体间的基因交流有意义(Green et al, 2010; Reich et al, 2010; Prufer et al, 2014), 同时也可以提供重组模式和适应性分化的信息(Kim & Rothschild, 2014).这些位点的识别对于揭示生殖隔离的遗传基础和找寻物种形成基因(speciation genes)有重要意义(Wu & Ting, 2004; Nosil & Schluter, 2011; Burri et al, 2015), 同时对建立针对濒危物种的保护策略有应用价值(der Sarkissian et al, 2015).基于基因组水平的滑动窗(sliding window)分析可以追溯不同染色体区段的祖先状态, 上面提到过的D-statistics就是一个很好的工具(Kronforst et al, 2013; Smith & Kronforst, 2013). ...
Integrating phylogenomic and morphological data to assess candidate species-delimitation models in brown and red-bellied snakes (Storeria).
1
2016
... 大量的调查分析表明, 和生殖隔离密切相关的性状更多地表现出超亲分离, 比如植物的环境适应性和开花物候性状、动物的生活史和行为性状(Rieseberg et al, 1999).总之, 杂种和亲本种间在表型的变异模式上有联系, 但这种联系还没有确定的模式可依.由此可见, 表型在揭示杂交是否存在或者确定亲本来源上有很多限制, 它无法提供确切的依据.但表型指标甚至仅仅是形态指标往往也能提供非常有价值的信息, 比如将它们与基因组水平的标记结合起来可以为物种的界定提供重要信息(Pyron et al, 2016).笔者亲历了一个体现形态数据重要性的实例. 在几十年前对松属(Pinus)的修订中, 我国的分类学家就依据形态特征猜测高山松(Pinus densata)要么来自云南松(P. yunnanensis)和油松(P. tabuliformis)的杂交, 要么作为祖先种分化为另外两种松树(吴中伦, 1956).之后, 研究人员开展了形态、生殖、物候、解剖、幼苗萌发、人工杂交、多重分子标记的群体遗传等多方面的研究, 已有的证据支持了前一个推测(Wang XR et al, 1990, 2001; Wang & Szmidt, 1994; Song et al, 2002, 2003; Ma et al, 2006; Mao et al, 2009; Wang BS et al, 2011; Gao et al, 2012; 张立沙等, 2012; 梁冬等, 2013; Xing et al, 2014; Zhao et al, 2014). ...
The genetic prehistory of the new world Arctic.
1
2014
... 重组图(admixture graph)是一个允许类群间基因交流的系统发育树的拓展.基于上面提到的F-statistics就可以构建这样的图, 目前有几种工具可以利用.MixMapper (Lipson et al, 2014)是一个半自动的工具.它首先基于由一对明显没有杂交的群体间等位基因频率(F2)求算的遗传聚类生成邻接树(neighbor-joining tree), 然后在允许基因流的情况下把剩下的群体添加到图上来(Lipson et al, 2013, 2014).TreeMix (Pickrell & Pritchard, 2012)实现了与MixMapper在理论上类似的算法.在假定重组或者基因流个数的情况下, 它可以全自动地计算出重组图.不同的重组次数的假定会对结果带来极大影响, 选取时需谨慎.上面提到的ADMIXTOOLS软件(Patterson et al, 2012)中也提供了类似的工具.但这里的重组图工具更加稳健, 因为它提供了假设中的重组模式是否和数据相符的检验.重组图的手段已经在人类遗传领域广泛应用, 曾被用来揭示北美原著民的遗传组成和来源(Raghavan et al, 2014b)、丹尼索瓦人(Denisovans)对现代人群的遗传贡献(Meyer et al, 2012)、新世界寒带地区的人类定居历史(Raghavan et al, 2014a). ...
Upper Palaeolithic Siberian genome reveals dual ancestry of native Americans.
1
2014
... 重组图(admixture graph)是一个允许类群间基因交流的系统发育树的拓展.基于上面提到的F-statistics就可以构建这样的图, 目前有几种工具可以利用.MixMapper (Lipson et al, 2014)是一个半自动的工具.它首先基于由一对明显没有杂交的群体间等位基因频率(F2)求算的遗传聚类生成邻接树(neighbor-joining tree), 然后在允许基因流的情况下把剩下的群体添加到图上来(Lipson et al, 2013, 2014).TreeMix (Pickrell & Pritchard, 2012)实现了与MixMapper在理论上类似的算法.在假定重组或者基因流个数的情况下, 它可以全自动地计算出重组图.不同的重组次数的假定会对结果带来极大影响, 选取时需谨慎.上面提到的ADMIXTOOLS软件(Patterson et al, 2012)中也提供了类似的工具.但这里的重组图工具更加稳健, 因为它提供了假设中的重组模式是否和数据相符的检验.重组图的手段已经在人类遗传领域广泛应用, 曾被用来揭示北美原著民的遗传组成和来源(Raghavan et al, 2014b)、丹尼索瓦人(Denisovans)对现代人群的遗传贡献(Meyer et al, 2012)、新世界寒带地区的人类定居历史(Raghavan et al, 2014a). ...
... 上面提到的主成分分析、聚类分析(比如基于structure和admixture的分析)可以用来澄清群体遗传结构, 并对杂交的存在提供全局性的线索(那些看似杂交的迹象可能由杂交以外的其他因素导致), 但它们都没有提供杂交是否存在的检验.比如, 与距离关联的分化(isolation by distance)可以产生在PCA上的梯度变异.基于structure和admixture的结果也很难做出对群体历史的推算, 因为它们没有对特定的群体历史模型进行检验, 而是简单地假定抽样群体都是从某特定群体快速辐射分化而来.Patterson等(2012)综合已有工作(Reich et al, 2009; Green et al, 2010; Durand et al, 2011; Moorjani et al, 2011), 归纳出了几个针对群体间杂交历史的检验和相应的参数, 并提供了实现有关计算的软件包(ADMIXTOOLS).这些参数和检验包括1个三群体检验(f3-statistic, the three-population test)和2个四群体检验(D-statistics或者ABBA-BABA test, 以及f4- statistics或F4-ratio).这里的f3-statistic和f4-statistics也被统称为F-statistics. ...
Genetic history of an archaic hominin group from Denisova Cave in Siberia.
1
2010
... 与杂交关联的是不同特征的基因组间的重组, 这种重组会带来一个嵌合的基因组, 在嵌合的基因组上不同的区域体现着不同的进化历程.识别这些位点不但对澄清种间或群体间的基因交流有意义(Green et al, 2010; Reich et al, 2010; Prufer et al, 2014), 同时也可以提供重组模式和适应性分化的信息(Kim & Rothschild, 2014).这些位点的识别对于揭示生殖隔离的遗传基础和找寻物种形成基因(speciation genes)有重要意义(Wu & Ting, 2004; Nosil & Schluter, 2011; Burri et al, 2015), 同时对建立针对濒危物种的保护策略有应用价值(der Sarkissian et al, 2015).基于基因组水平的滑动窗(sliding window)分析可以追溯不同染色体区段的祖先状态, 上面提到过的D-statistics就是一个很好的工具(Kronforst et al, 2013; Smith & Kronforst, 2013). ...
Long identical multispecies elements in plant and animal genomes
2
2012
... 尽管目标富集测序技术相对成熟, 但目前的应用较多地针对人类疾病研究, 在系统发育分析中的应用仍在发展.探针开发是一个重要环节, 从转录组或基因组开发探针的工具和流程已有不少报道(Mayer et al, 2016; Pavy et al, 2016; Schmickl et al, 2016).超保守序列(ultraconserved elements)也是开发捕获探针的重要来源, 目前哺乳动物(Bejerano et al, 2004; Reneker et al, 2012)、鸟类(Mccormack & Al, 2011)、两栖爬行类(Crawford et al, 2012)、昆虫(Branstetter et al, 2017)、高等植物(Freeling et al, 2009; Reneker et al, 2012)上的超保守序列已有报道.该技术还有一个特点, 就是通过对长目标序列多个不同区域设计探针, 可以实现对整条序列的捕获测序.这突破了目前流行的新一代测序的读长较短的限制.但这对数据分析也造成了问题, 因为目前流行的系统发育分析软件对大量长片段进行分析时计算速度往往很慢, 比如BEST (Edwards et al, 2007)和*BEST (Heled & Drummond, 2010). ...
... ; Reneker et al, 2012)上的超保守序列已有报道.该技术还有一个特点, 就是通过对长目标序列多个不同区域设计探针, 可以实现对整条序列的捕获测序.这突破了目前流行的新一代测序的读长较短的限制.但这对数据分析也造成了问题, 因为目前流行的系统发育分析软件对大量长片段进行分析时计算速度往往很慢, 比如BEST (Edwards et al, 2007)和*BEST (Heled & Drummond, 2010). ...
Transgressive segregation, adaptation and speciation.
2
1999
... 在分子标记技术出现前, 对杂交的描述大都依赖表型分析.表型指个体在包括形态、行为、物候、生殖、生理、生活史、次生代谢等各方面的表现.F1代杂种往往体现出两个亲本都不具备的杂种优势; 不论低世代还是高世代杂种, 除了受到亲本遗传物质比例的影响之外, 它们往往在一些性状上表现出居中的表型, 同时都普遍在个别性状上体现出超亲分离(transgressive segregation).在调查的171个研究实例中, 155个(91%)有超亲分离存在; 在分析的1,229个性状中, 有44%显示了超亲分离, 其中58%的植物性状和35%的动物性状显示了超亲分离(Rieseberg et al, 1999).杂种显示超亲分离与杂交本身有直接关系(Rieseberg et al, 2003). ...
... 大量的调查分析表明, 和生殖隔离密切相关的性状更多地表现出超亲分离, 比如植物的环境适应性和开花物候性状、动物的生活史和行为性状(Rieseberg et al, 1999).总之, 杂种和亲本种间在表型的变异模式上有联系, 但这种联系还没有确定的模式可依.由此可见, 表型在揭示杂交是否存在或者确定亲本来源上有很多限制, 它无法提供确切的依据.但表型指标甚至仅仅是形态指标往往也能提供非常有价值的信息, 比如将它们与基因组水平的标记结合起来可以为物种的界定提供重要信息(Pyron et al, 2016).笔者亲历了一个体现形态数据重要性的实例. 在几十年前对松属(Pinus)的修订中, 我国的分类学家就依据形态特征猜测高山松(Pinus densata)要么来自云南松(P. yunnanensis)和油松(P. tabuliformis)的杂交, 要么作为祖先种分化为另外两种松树(吴中伦, 1956).之后, 研究人员开展了形态、生殖、物候、解剖、幼苗萌发、人工杂交、多重分子标记的群体遗传等多方面的研究, 已有的证据支持了前一个推测(Wang XR et al, 1990, 2001; Wang & Szmidt, 1994; Song et al, 2002, 2003; Ma et al, 2006; Mao et al, 2009; Wang BS et al, 2011; Gao et al, 2012; 张立沙等, 2012; 梁冬等, 2013; Xing et al, 2014; Zhao et al, 2014). ...
The genetic architecture necessary for transgressive segregation is common in both natural and domesticated populations.
1
2003
... 在分子标记技术出现前, 对杂交的描述大都依赖表型分析.表型指个体在包括形态、行为、物候、生殖、生理、生活史、次生代谢等各方面的表现.F1代杂种往往体现出两个亲本都不具备的杂种优势; 不论低世代还是高世代杂种, 除了受到亲本遗传物质比例的影响之外, 它们往往在一些性状上表现出居中的表型, 同时都普遍在个别性状上体现出超亲分离(transgressive segregation).在调查的171个研究实例中, 155个(91%)有超亲分离存在; 在分析的1,229个性状中, 有44%显示了超亲分离, 其中58%的植物性状和35%的动物性状显示了超亲分离(Rieseberg et al, 1999).杂种显示超亲分离与杂交本身有直接关系(Rieseberg et al, 2003). ...
ABC inference of multi-population divergence with admixture from unphased population genomic data.
1
2014
... 基于最大似然算法的策略往往很复杂, 需要大量的计算.近似贝叶斯算法(approximate Bayesian computation, ABC)不需要计算似然值, 它提供了一个检验各种复杂进化过程是否存在的平台(Beaumont, 2010).近似贝叶斯计算中, 首先对待检测过程依据一定的期望建立多重模拟, 然后将真实数据和模拟产生的数据进行比较, 再通过选取那些与真实数据最类似的模型来实现对待检验假设的推断.近似贝叶斯计算需要完成数据模拟、统计计算、模型比较等步骤, 目前有多种针对性的工具可以利用, 可参考一些重要综述文章(Csilléry et al, 2010; Sunnåker et al, 2013; Lintusaari et al, 2016).DIYABC (Cornuet et al, 2014)、ABCtoolbox (Wegmann et al, 2010)和abc (Csilléry et al, 2011)等是通常用来实现ABC计算的平台.利用基因组水平未定向数据和ABC算法揭示杂交事件存在的一个研究实例来自针对胡蜂(Biorhiza pallida)的工作(Robinson et al, 2014). ...
Detecting and overcoming systematic errors in genome-scale phylogenies.
1
2007
... 模型初选后, 应该用恰当的检验来评判选定模型的可靠性.这方面有一系列的工具可以利用, 比如位点内或位点间重组(Mcguire et al, 1997; Kosakovsky et al, 2006; Martin et al, 2015)、各种选择(Delport et al, 2010)、杂交(Yu et al, 2011)、时序性进化速率差异(heterotachy) (Pagel & Meade, 2008)和模型总体上的配合性(Albert & Schluter, 2005; Abby et al, 2012; Ackermann et al, 2016)等.如果初选模型不适合, 那么应该及时调整选用更恰当的模型.如果位点间基因树冲突, 那么可考虑选用应对杂交或者不完全谱系分离的模型, 而不是仍然将序列合并使用, 或者把不相配合的部分数据删除.当然还是要删除那些体现强烈选择印记的、变异饱和的(saturated) (Castresana, 2000; Rodríguezezpeleta et al, 2007; Gnirke et al, 2009)和包含有缺失值(missing)或有缺失变异(deletion)的位点(Lemmon et al, 2009). ...
Ancestry inference in complex admixtures via variable- length Markov chain linkage models.
1
2013
... 已经有一些估算位点祖先状态的计算工具, Padhukasahasram (2014)就进行了较为详尽的综述.一般来说, 这些计算工具需要借助隐马尔科夫模型(hidden Markov models, HMMs)来确定不同亲本来源的基因组区段的界限.HAPMIX软件可以利用未定相(unphased)的基因型数据确定杂种个体特定位点的祖先来源(Price et al, 2009).CRFs应用一个广义隐马尔科夫模型(generalized hidden Markov model), 借助训练集数据实现了对祖先来源的估算(Sankararaman et al, 2012).实现这类分析的软件还包括: SABER (Tang et al, 2006)、HAPA (Sundquist et al, 2008)、LAMP (Sankararaman et al, 2008)、LAMP- LD/LAMP-HAP (Baran et al, 2012)、WINPOP (Pasaniuc et al, 2009)、SupportMix (Omberg et al, 2012)、ASPCA (Moreno-Estrada et al, 2013)、ALLOY (Rodriguez et al, 2013)、RFMix (Maples et al, 2013)、Lanc- CSV (Brown & Pasaniuc, 2014)和EILA (Yang et al, 2013).EILA没有对输入数据位点间连锁平衡的前提假设, 且比LAMP和HAPMIX更准确, 运行速度也不慢.Lanc-CSV实现了经典算法, 但对超大样本数据有速度上的优势. ...
Focus on next-generation sequencing data analysis.
The date of interbreeding between Neandertals and modern humans.
1
2012
... 已经有一些估算位点祖先状态的计算工具, Padhukasahasram (2014)就进行了较为详尽的综述.一般来说, 这些计算工具需要借助隐马尔科夫模型(hidden Markov models, HMMs)来确定不同亲本来源的基因组区段的界限.HAPMIX软件可以利用未定相(unphased)的基因型数据确定杂种个体特定位点的祖先来源(Price et al, 2009).CRFs应用一个广义隐马尔科夫模型(generalized hidden Markov model), 借助训练集数据实现了对祖先来源的估算(Sankararaman et al, 2012).实现这类分析的软件还包括: SABER (Tang et al, 2006)、HAPA (Sundquist et al, 2008)、LAMP (Sankararaman et al, 2008)、LAMP- LD/LAMP-HAP (Baran et al, 2012)、WINPOP (Pasaniuc et al, 2009)、SupportMix (Omberg et al, 2012)、ASPCA (Moreno-Estrada et al, 2013)、ALLOY (Rodriguez et al, 2013)、RFMix (Maples et al, 2013)、Lanc- CSV (Brown & Pasaniuc, 2014)和EILA (Yang et al, 2013).EILA没有对输入数据位点间连锁平衡的前提假设, 且比LAMP和HAPMIX更准确, 运行速度也不慢.Lanc-CSV实现了经典算法, 但对超大样本数据有速度上的优势. ...
The genomic landscape of Neanderthal ancestry in present-day humans.
1
2014
... 这个技术的实现是显而易见的.对于已有参考基因组、基因组大小合适的类群, 全基因组测序(更多的是基因组重测序, genome resequencing)应用已经很常见了, 尤其是对近缘种间或种内群体分化的研究中, 其优势更加明显.人类的1,000 Genomes计划(http://www.internationalgenome.org/)(The Genomes Project Consortium et al, 2010)和植物中拟南芥(Arabidopsis thaliana)的1,001 Genomes计划(http://1001genomes.org/) (Weigel & Mott, 2009)都为我们作了很好的示范.值得一提的是, 基因组水平的证据表明种间遗传交换参与了人(Patterson et al, 2012; Hellenthal et al, 2014; Lazaridis et al, 2014; Sankararaman et al, 2014; Ackermann et al, 2016)和拟南芥(Stenz et al, 2015; Novikova et al, 2016)的起源和分化. ...
Estimating local ancestry in admixed populations.
1
2008
... 已经有一些估算位点祖先状态的计算工具, Padhukasahasram (2014)就进行了较为详尽的综述.一般来说, 这些计算工具需要借助隐马尔科夫模型(hidden Markov models, HMMs)来确定不同亲本来源的基因组区段的界限.HAPMIX软件可以利用未定相(unphased)的基因型数据确定杂种个体特定位点的祖先来源(Price et al, 2009).CRFs应用一个广义隐马尔科夫模型(generalized hidden Markov model), 借助训练集数据实现了对祖先来源的估算(Sankararaman et al, 2012).实现这类分析的软件还包括: SABER (Tang et al, 2006)、HAPA (Sundquist et al, 2008)、LAMP (Sankararaman et al, 2008)、LAMP- LD/LAMP-HAP (Baran et al, 2012)、WINPOP (Pasaniuc et al, 2009)、SupportMix (Omberg et al, 2012)、ASPCA (Moreno-Estrada et al, 2013)、ALLOY (Rodriguez et al, 2013)、RFMix (Maples et al, 2013)、Lanc- CSV (Brown & Pasaniuc, 2014)和EILA (Yang et al, 2013).EILA没有对输入数据位点间连锁平衡的前提假设, 且比LAMP和HAPMIX更准确, 运行速度也不慢.Lanc-CSV实现了经典算法, 但对超大样本数据有速度上的优势. ...
Phylogenetic marker development for target enrichment from transcriptome and genome skim data: the pipeline and its application in southern African Oxalis (Oxalidaceae).
1
2016
... 尽管目标富集测序技术相对成熟, 但目前的应用较多地针对人类疾病研究, 在系统发育分析中的应用仍在发展.探针开发是一个重要环节, 从转录组或基因组开发探针的工具和流程已有不少报道(Mayer et al, 2016; Pavy et al, 2016; Schmickl et al, 2016).超保守序列(ultraconserved elements)也是开发捕获探针的重要来源, 目前哺乳动物(Bejerano et al, 2004; Reneker et al, 2012)、鸟类(Mccormack & Al, 2011)、两栖爬行类(Crawford et al, 2012)、昆虫(Branstetter et al, 2017)、高等植物(Freeling et al, 2009; Reneker et al, 2012)上的超保守序列已有报道.该技术还有一个特点, 就是通过对长目标序列多个不同区域设计探针, 可以实现对整条序列的捕获测序.这突破了目前流行的新一代测序的读长较短的限制.但这对数据分析也造成了问题, 因为目前流行的系统发育分析软件对大量长片段进行分析时计算速度往往很慢, 比如BEST (Edwards et al, 2007)和*BEST (Heled & Drummond, 2010). ...
Introduction: extent, processes and evolutionary impact of interspecific hybridization in animals.
1
2008
... 生物类群中杂交的普遍存在获得了越来越多的认可.然而, 相对于复杂的生物多样性, 清晰描述的杂交事件仍显得非常少.实际上, 作为一个重要的进化过程和生物多样性维持机制, 杂交受到的关注还远远不够.据估算, 有25%的高等植物物种(Mallet, 2005)和10%的动物物种(Schwenk et al, 2008)间存在着种间杂交; 此外, 在真菌(Nelson, 1963; Depotter et al, 2016)、细菌(Duncan et al, 1989; Cohan & Kane, 2001; Cohan, 2002; Earl et al, 2008)、病毒(Worobey & Holmes, 1999; Bujarski, 2013; Lefeuvre & Moriones, 2015; Pérez-Losada et al, 2015; Su et al, 2016)的基因组进化中, 杂交普遍存在.以植物为例, Flora of China共记载我国3万余种植物(Wu et al, 2014), 按25%计算, 我们推测其中9,000种植物间存在杂交.同时, 日益加剧的全球气候变化和人类活动将对生物多样性的分布特征产生显著影响, 进而改变种间关系, 导致原已形成的种间生殖隔离改变或降低, 进一步增加杂交的机会(Stenz et al, 2015; Novikova et al, 2016; Vallejo- Marin & Hiscock, 2016).相较于上面的估算, 得到确认的植物杂交案例非常稀少, 我们认为可能不足1%.因此, 要全面澄清杂交产生的机制、杂交对多样性形成和维持的影响及其进化生物学意义还为时尚早.目前已经开展或正在进行的工作所覆盖的杂交事件, 估计不超过总量的1‰.检测杂交及评估其发生的范围和机制, 将成为未来生物多样性监测、管理和保护中不可缺少的组成部分.在后志书时代的今天, 检测杂交等遗传交换事件的存在, 进而揭示它对生物多样性形成和维持的重要价值, 是我们面临的重要挑战. ...
Bioinformatic processing of RAD- seq data dramatically impacts downstream population genetic inference.
1
2016
... 相对传统技术来说, 这些分子标记技术的优势十分明显, 尽管数据处理中的某些具体环节还在发展, 但成熟的技术流程和相应的软件并不难获得.测序仪输出的原始数据转换、短读(short read)长数据质控、短读长数据的比对、转录组或基因组的组装、单碱基突变或单倍型获取、各步骤质量评估和数据筛选等重要环节的分析对一般的系统发育和群体遗传学的研究团队并不难实现, 有大量的高质量技术指南可以参考(DePristo et al, 2011; Nielsen et al, 2011).对某一种特定分子标记技术的分析策略也有不少参考, 比如RAD测序方面(Hapke & Thiele, 2016; Kim et al, 2016; McKinney et al, 2016; Shafer et al, 2016; Torkamaneh et al, 2016, 2017). ...
A versatile and highly efficient toolkit including 102 nuclear markers for vertebrate phylogenomics, tested by resolving the higher level relationships of the caudata.
Cytoplasmic composition in Pinus densata and population establishment of the diploid hybrid pine.
1
2003
... 大量的调查分析表明, 和生殖隔离密切相关的性状更多地表现出超亲分离, 比如植物的环境适应性和开花物候性状、动物的生活史和行为性状(Rieseberg et al, 1999).总之, 杂种和亲本种间在表型的变异模式上有联系, 但这种联系还没有确定的模式可依.由此可见, 表型在揭示杂交是否存在或者确定亲本来源上有很多限制, 它无法提供确切的依据.但表型指标甚至仅仅是形态指标往往也能提供非常有价值的信息, 比如将它们与基因组水平的标记结合起来可以为物种的界定提供重要信息(Pyron et al, 2016).笔者亲历了一个体现形态数据重要性的实例. 在几十年前对松属(Pinus)的修订中, 我国的分类学家就依据形态特征猜测高山松(Pinus densata)要么来自云南松(P. yunnanensis)和油松(P. tabuliformis)的杂交, 要么作为祖先种分化为另外两种松树(吴中伦, 1956).之后, 研究人员开展了形态、生殖、物候、解剖、幼苗萌发、人工杂交、多重分子标记的群体遗传等多方面的研究, 已有的证据支持了前一个推测(Wang XR et al, 1990, 2001; Wang & Szmidt, 1994; Song et al, 2002, 2003; Ma et al, 2006; Mao et al, 2009; Wang BS et al, 2011; Gao et al, 2012; 张立沙等, 2012; 梁冬等, 2013; Xing et al, 2014; Zhao et al, 2014). ...
Maternal lineages of Pinus densata, a diploid, hybrid.
1
2002
... 大量的调查分析表明, 和生殖隔离密切相关的性状更多地表现出超亲分离, 比如植物的环境适应性和开花物候性状、动物的生活史和行为性状(Rieseberg et al, 1999).总之, 杂种和亲本种间在表型的变异模式上有联系, 但这种联系还没有确定的模式可依.由此可见, 表型在揭示杂交是否存在或者确定亲本来源上有很多限制, 它无法提供确切的依据.但表型指标甚至仅仅是形态指标往往也能提供非常有价值的信息, 比如将它们与基因组水平的标记结合起来可以为物种的界定提供重要信息(Pyron et al, 2016).笔者亲历了一个体现形态数据重要性的实例. 在几十年前对松属(Pinus)的修订中, 我国的分类学家就依据形态特征猜测高山松(Pinus densata)要么来自云南松(P. yunnanensis)和油松(P. tabuliformis)的杂交, 要么作为祖先种分化为另外两种松树(吴中伦, 1956).之后, 研究人员开展了形态、生殖、物候、解剖、幼苗萌发、人工杂交、多重分子标记的群体遗传等多方面的研究, 已有的证据支持了前一个推测(Wang XR et al, 1990, 2001; Wang & Szmidt, 1994; Song et al, 2002, 2003; Ma et al, 2006; Mao et al, 2009; Wang BS et al, 2011; Gao et al, 2012; 张立沙等, 2012; 梁冬等, 2013; Xing et al, 2014; Zhao et al, 2014). ...
Hybridization and hybrid speciation under global change.
1
2016
... 生物类群中杂交的普遍存在获得了越来越多的认可.然而, 相对于复杂的生物多样性, 清晰描述的杂交事件仍显得非常少.实际上, 作为一个重要的进化过程和生物多样性维持机制, 杂交受到的关注还远远不够.据估算, 有25%的高等植物物种(Mallet, 2005)和10%的动物物种(Schwenk et al, 2008)间存在着种间杂交; 此外, 在真菌(Nelson, 1963; Depotter et al, 2016)、细菌(Duncan et al, 1989; Cohan & Kane, 2001; Cohan, 2002; Earl et al, 2008)、病毒(Worobey & Holmes, 1999; Bujarski, 2013; Lefeuvre & Moriones, 2015; Pérez-Losada et al, 2015; Su et al, 2016)的基因组进化中, 杂交普遍存在.以植物为例, Flora of China共记载我国3万余种植物(Wu et al, 2014), 按25%计算, 我们推测其中9,000种植物间存在杂交.同时, 日益加剧的全球气候变化和人类活动将对生物多样性的分布特征产生显著影响, 进而改变种间关系, 导致原已形成的种间生殖隔离改变或降低, 进一步增加杂交的机会(Stenz et al, 2015; Novikova et al, 2016; Vallejo- Marin & Hiscock, 2016).相较于上面的估算, 得到确认的植物杂交案例非常稀少, 我们认为可能不足1%.因此, 要全面澄清杂交产生的机制、杂交对多样性形成和维持的影响及其进化生物学意义还为时尚早.目前已经开展或正在进行的工作所覆盖的杂交事件, 估计不超过总量的1‰.检测杂交及评估其发生的范围和机制, 将成为未来生物多样性监测、管理和保护中不可缺少的组成部分.在后志书时代的今天, 检测杂交等遗传交换事件的存在, 进而揭示它对生物多样性形成和维持的重要价值, 是我们面临的重要挑战. ...
SNP discovery and allele frequency estimation by deep sequencing of reduced representation libraries.
1
2008
... 这类技术都包含一个重要步骤, 即对目标样本基因组进行限制性内切酶酶切, 有些还包含对酶切产物片段大小的筛选(Baird et al, 2008; van Tassell et al, 2008; Kim et al, 2016).酶切过程中, 可用单一的内切酶, 也可用两个内切酶的组合(Peterson et al, 2012); 酶切的特征可能是甲基化敏感的、甲基化不敏感的或IIB型限制性核酸内切酶(Wang et al, 2012).酶的组合和酶切片段筛选给了这类技术极大的灵活性, 加上样本标签后可以实现各种组合的混样测序, 这类技术的潜力巨大.基于类似的简化原理, 已有大量特定技术衍生出来(如reduced-representation library sequencing (RRL), restriction-site- associated DNA sequencing (RAD), genotyping by sequencing (GBS)), 有不少综述性论文对它们进行了详细介绍(Davey et al, 2011; Andrews et al, 2016).该分子标记技术的特征参见图1. ...
Colonization of the Tibetan Plateau by the homoploid hybrid pine Pinus densata.
1
2011
... 大量的调查分析表明, 和生殖隔离密切相关的性状更多地表现出超亲分离, 比如植物的环境适应性和开花物候性状、动物的生活史和行为性状(Rieseberg et al, 1999).总之, 杂种和亲本种间在表型的变异模式上有联系, 但这种联系还没有确定的模式可依.由此可见, 表型在揭示杂交是否存在或者确定亲本来源上有很多限制, 它无法提供确切的依据.但表型指标甚至仅仅是形态指标往往也能提供非常有价值的信息, 比如将它们与基因组水平的标记结合起来可以为物种的界定提供重要信息(Pyron et al, 2016).笔者亲历了一个体现形态数据重要性的实例. 在几十年前对松属(Pinus)的修订中, 我国的分类学家就依据形态特征猜测高山松(Pinus densata)要么来自云南松(P. yunnanensis)和油松(P. tabuliformis)的杂交, 要么作为祖先种分化为另外两种松树(吴中伦, 1956).之后, 研究人员开展了形态、生殖、物候、解剖、幼苗萌发、人工杂交、多重分子标记的群体遗传等多方面的研究, 已有的证据支持了前一个推测(Wang XR et al, 1990, 2001; Wang & Szmidt, 1994; Song et al, 2002, 2003; Ma et al, 2006; Mao et al, 2009; Wang BS et al, 2011; Gao et al, 2012; 张立沙等, 2012; 梁冬等, 2013; Xing et al, 2014; Zhao et al, 2014). ...
2b-RAD: a simple and flexible method for genome-wide genotyping.
1
2012
... 这类技术都包含一个重要步骤, 即对目标样本基因组进行限制性内切酶酶切, 有些还包含对酶切产物片段大小的筛选(Baird et al, 2008; van Tassell et al, 2008; Kim et al, 2016).酶切过程中, 可用单一的内切酶, 也可用两个内切酶的组合(Peterson et al, 2012); 酶切的特征可能是甲基化敏感的、甲基化不敏感的或IIB型限制性核酸内切酶(Wang et al, 2012).酶的组合和酶切片段筛选给了这类技术极大的灵活性, 加上样本标签后可以实现各种组合的混样测序, 这类技术的潜力巨大.基于类似的简化原理, 已有大量特定技术衍生出来(如reduced-representation library sequencing (RRL), restriction-site- associated DNA sequencing (RAD), genotyping by sequencing (GBS)), 有不少综述性论文对它们进行了详细介绍(Davey et al, 2011; Andrews et al, 2016).该分子标记技术的特征参见图1. ...
Hybridization and chloroplast DNA variation in a Pinus species complex from Asia.
1
1994
... 大量的调查分析表明, 和生殖隔离密切相关的性状更多地表现出超亲分离, 比如植物的环境适应性和开花物候性状、动物的生活史和行为性状(Rieseberg et al, 1999).总之, 杂种和亲本种间在表型的变异模式上有联系, 但这种联系还没有确定的模式可依.由此可见, 表型在揭示杂交是否存在或者确定亲本来源上有很多限制, 它无法提供确切的依据.但表型指标甚至仅仅是形态指标往往也能提供非常有价值的信息, 比如将它们与基因组水平的标记结合起来可以为物种的界定提供重要信息(Pyron et al, 2016).笔者亲历了一个体现形态数据重要性的实例. 在几十年前对松属(Pinus)的修订中, 我国的分类学家就依据形态特征猜测高山松(Pinus densata)要么来自云南松(P. yunnanensis)和油松(P. tabuliformis)的杂交, 要么作为祖先种分化为另外两种松树(吴中伦, 1956).之后, 研究人员开展了形态、生殖、物候、解剖、幼苗萌发、人工杂交、多重分子标记的群体遗传等多方面的研究, 已有的证据支持了前一个推测(Wang XR et al, 1990, 2001; Wang & Szmidt, 1994; Song et al, 2002, 2003; Ma et al, 2006; Mao et al, 2009; Wang BS et al, 2011; Gao et al, 2012; 张立沙等, 2012; 梁冬等, 2013; Xing et al, 2014; Zhao et al, 2014). ...
Genetic composition and diploid hybrid speciation of a high mountain pine, Pinus densata, native to the Tibetan Plateau.
1
2001
... 大量的调查分析表明, 和生殖隔离密切相关的性状更多地表现出超亲分离, 比如植物的环境适应性和开花物候性状、动物的生活史和行为性状(Rieseberg et al, 1999).总之, 杂种和亲本种间在表型的变异模式上有联系, 但这种联系还没有确定的模式可依.由此可见, 表型在揭示杂交是否存在或者确定亲本来源上有很多限制, 它无法提供确切的依据.但表型指标甚至仅仅是形态指标往往也能提供非常有价值的信息, 比如将它们与基因组水平的标记结合起来可以为物种的界定提供重要信息(Pyron et al, 2016).笔者亲历了一个体现形态数据重要性的实例. 在几十年前对松属(Pinus)的修订中, 我国的分类学家就依据形态特征猜测高山松(Pinus densata)要么来自云南松(P. yunnanensis)和油松(P. tabuliformis)的杂交, 要么作为祖先种分化为另外两种松树(吴中伦, 1956).之后, 研究人员开展了形态、生殖、物候、解剖、幼苗萌发、人工杂交、多重分子标记的群体遗传等多方面的研究, 已有的证据支持了前一个推测(Wang XR et al, 1990, 2001; Wang & Szmidt, 1994; Song et al, 2002, 2003; Ma et al, 2006; Mao et al, 2009; Wang BS et al, 2011; Gao et al, 2012; 张立沙等, 2012; 梁冬等, 2013; Xing et al, 2014; Zhao et al, 2014). ...
Evolutionary analysis of Pinus densata Masters, a putative tertiary hybrid 1, allozyme variation.
1
1990
... 大量的调查分析表明, 和生殖隔离密切相关的性状更多地表现出超亲分离, 比如植物的环境适应性和开花物候性状、动物的生活史和行为性状(Rieseberg et al, 1999).总之, 杂种和亲本种间在表型的变异模式上有联系, 但这种联系还没有确定的模式可依.由此可见, 表型在揭示杂交是否存在或者确定亲本来源上有很多限制, 它无法提供确切的依据.但表型指标甚至仅仅是形态指标往往也能提供非常有价值的信息, 比如将它们与基因组水平的标记结合起来可以为物种的界定提供重要信息(Pyron et al, 2016).笔者亲历了一个体现形态数据重要性的实例. 在几十年前对松属(Pinus)的修订中, 我国的分类学家就依据形态特征猜测高山松(Pinus densata)要么来自云南松(P. yunnanensis)和油松(P. tabuliformis)的杂交, 要么作为祖先种分化为另外两种松树(吴中伦, 1956).之后, 研究人员开展了形态、生殖、物候、解剖、幼苗萌发、人工杂交、多重分子标记的群体遗传等多方面的研究, 已有的证据支持了前一个推测(Wang XR et al, 1990, 2001; Wang & Szmidt, 1994; Song et al, 2002, 2003; Ma et al, 2006; Mao et al, 2009; Wang BS et al, 2011; Gao et al, 2012; 张立沙等, 2012; 梁冬等, 2013; Xing et al, 2014; Zhao et al, 2014). ...
RNA-Seq: a revolutionary tool for transcriptomics.
1
2009
... 转录组测序(transcriptome sequencing)或者RNA测序(RNA sequencing)是针对基因组中的表达序列进行测序, 本质上也是一种简化基因组测序技术(Marioni et al, 2008; Morin et al, 2008; Wang et al, 2009).尽管这项技术通常被用于适应性进化和生态基因组学研究, 但已有的研究实例表明, 该技术获得的转录组序列可以用于重建系统发育和检测杂交的存在(Nabholz et al, 2011; Pease et al, 2016).此外, 该技术对发掘单碱基突变、进行群体遗传研究也很有吸引力, 因为它不仅可实现对复杂基因组的简化, 而且提供了来自直接与功能相关转录序列的信息.从转录组组装序列中寻找直系同源基因的计算工具已有报道, HaMStR (Ebersberger et al, 2009)是比较不错的一个.该分子标记技术的特征参见图1. ...
ABCtoolbox: a versatile toolkit for approximate Bayesian computations.
1
2010
... 基于最大似然算法的策略往往很复杂, 需要大量的计算.近似贝叶斯算法(approximate Bayesian computation, ABC)不需要计算似然值, 它提供了一个检验各种复杂进化过程是否存在的平台(Beaumont, 2010).近似贝叶斯计算中, 首先对待检测过程依据一定的期望建立多重模拟, 然后将真实数据和模拟产生的数据进行比较, 再通过选取那些与真实数据最类似的模型来实现对待检验假设的推断.近似贝叶斯计算需要完成数据模拟、统计计算、模型比较等步骤, 目前有多种针对性的工具可以利用, 可参考一些重要综述文章(Csilléry et al, 2010; Sunnåker et al, 2013; Lintusaari et al, 2016).DIYABC (Cornuet et al, 2014)、ABCtoolbox (Wegmann et al, 2010)和abc (Csilléry et al, 2011)等是通常用来实现ABC计算的平台.利用基因组水平未定向数据和ABC算法揭示杂交事件存在的一个研究实例来自针对胡蜂(Biorhiza pallida)的工作(Robinson et al, 2014). ...
The 1001 Genomes Project for Arabidopsis thaliana.
1
2009
... 这个技术的实现是显而易见的.对于已有参考基因组、基因组大小合适的类群, 全基因组测序(更多的是基因组重测序, genome resequencing)应用已经很常见了, 尤其是对近缘种间或种内群体分化的研究中, 其优势更加明显.人类的1,000 Genomes计划(http://www.internationalgenome.org/)(The Genomes Project Consortium et al, 2010)和植物中拟南芥(Arabidopsis thaliana)的1,001 Genomes计划(http://1001genomes.org/) (Weigel & Mott, 2009)都为我们作了很好的示范.值得一提的是, 基因组水平的证据表明种间遗传交换参与了人(Patterson et al, 2012; Hellenthal et al, 2014; Lazaridis et al, 2014; Sankararaman et al, 2014; Ackermann et al, 2016)和拟南芥(Stenz et al, 2015; Novikova et al, 2016)的起源和分化. ...
Evolutionary aspects of recombination in RNA viruses.
1
1999
... 生物类群中杂交的普遍存在获得了越来越多的认可.然而, 相对于复杂的生物多样性, 清晰描述的杂交事件仍显得非常少.实际上, 作为一个重要的进化过程和生物多样性维持机制, 杂交受到的关注还远远不够.据估算, 有25%的高等植物物种(Mallet, 2005)和10%的动物物种(Schwenk et al, 2008)间存在着种间杂交; 此外, 在真菌(Nelson, 1963; Depotter et al, 2016)、细菌(Duncan et al, 1989; Cohan & Kane, 2001; Cohan, 2002; Earl et al, 2008)、病毒(Worobey & Holmes, 1999; Bujarski, 2013; Lefeuvre & Moriones, 2015; Pérez-Losada et al, 2015; Su et al, 2016)的基因组进化中, 杂交普遍存在.以植物为例, Flora of China共记载我国3万余种植物(Wu et al, 2014), 按25%计算, 我们推测其中9,000种植物间存在杂交.同时, 日益加剧的全球气候变化和人类活动将对生物多样性的分布特征产生显著影响, 进而改变种间关系, 导致原已形成的种间生殖隔离改变或降低, 进一步增加杂交的机会(Stenz et al, 2015; Novikova et al, 2016; Vallejo- Marin & Hiscock, 2016).相较于上面的估算, 得到确认的植物杂交案例非常稀少, 我们认为可能不足1%.因此, 要全面澄清杂交产生的机制、杂交对多样性形成和维持的影响及其进化生物学意义还为时尚早.目前已经开展或正在进行的工作所覆盖的杂交事件, 估计不超过总量的1‰.检测杂交及评估其发生的范围和机制, 将成为未来生物多样性监测、管理和保护中不可缺少的组成部分.在后志书时代的今天, 检测杂交等遗传交换事件的存在, 进而揭示它对生物多样性形成和维持的重要价值, 是我们面临的重要挑战. ...
中国松属的分类与分布
1
1956
... 大量的调查分析表明, 和生殖隔离密切相关的性状更多地表现出超亲分离, 比如植物的环境适应性和开花物候性状、动物的生活史和行为性状(Rieseberg et al, 1999).总之, 杂种和亲本种间在表型的变异模式上有联系, 但这种联系还没有确定的模式可依.由此可见, 表型在揭示杂交是否存在或者确定亲本来源上有很多限制, 它无法提供确切的依据.但表型指标甚至仅仅是形态指标往往也能提供非常有价值的信息, 比如将它们与基因组水平的标记结合起来可以为物种的界定提供重要信息(Pyron et al, 2016).笔者亲历了一个体现形态数据重要性的实例. 在几十年前对松属(Pinus)的修订中, 我国的分类学家就依据形态特征猜测高山松(Pinus densata)要么来自云南松(P. yunnanensis)和油松(P. tabuliformis)的杂交, 要么作为祖先种分化为另外两种松树(吴中伦, 1956).之后, 研究人员开展了形态、生殖、物候、解剖、幼苗萌发、人工杂交、多重分子标记的群体遗传等多方面的研究, 已有的证据支持了前一个推测(Wang XR et al, 1990, 2001; Wang & Szmidt, 1994; Song et al, 2002, 2003; Ma et al, 2006; Mao et al, 2009; Wang BS et al, 2011; Gao et al, 2012; 张立沙等, 2012; 梁冬等, 2013; Xing et al, 2014; Zhao et al, 2014). ...
中国松属的分类与分布
1
1956
... 大量的调查分析表明, 和生殖隔离密切相关的性状更多地表现出超亲分离, 比如植物的环境适应性和开花物候性状、动物的生活史和行为性状(Rieseberg et al, 1999).总之, 杂种和亲本种间在表型的变异模式上有联系, 但这种联系还没有确定的模式可依.由此可见, 表型在揭示杂交是否存在或者确定亲本来源上有很多限制, 它无法提供确切的依据.但表型指标甚至仅仅是形态指标往往也能提供非常有价值的信息, 比如将它们与基因组水平的标记结合起来可以为物种的界定提供重要信息(Pyron et al, 2016).笔者亲历了一个体现形态数据重要性的实例. 在几十年前对松属(Pinus)的修订中, 我国的分类学家就依据形态特征猜测高山松(Pinus densata)要么来自云南松(P. yunnanensis)和油松(P. tabuliformis)的杂交, 要么作为祖先种分化为另外两种松树(吴中伦, 1956).之后, 研究人员开展了形态、生殖、物候、解剖、幼苗萌发、人工杂交、多重分子标记的群体遗传等多方面的研究, 已有的证据支持了前一个推测(Wang XR et al, 1990, 2001; Wang & Szmidt, 1994; Song et al, 2002, 2003; Ma et al, 2006; Mao et al, 2009; Wang BS et al, 2011; Gao et al, 2012; 张立沙等, 2012; 梁冬等, 2013; Xing et al, 2014; Zhao et al, 2014). ...
Genes and speciation.
1
2004
... 与杂交关联的是不同特征的基因组间的重组, 这种重组会带来一个嵌合的基因组, 在嵌合的基因组上不同的区域体现着不同的进化历程.识别这些位点不但对澄清种间或群体间的基因交流有意义(Green et al, 2010; Reich et al, 2010; Prufer et al, 2014), 同时也可以提供重组模式和适应性分化的信息(Kim & Rothschild, 2014).这些位点的识别对于揭示生殖隔离的遗传基础和找寻物种形成基因(speciation genes)有重要意义(Wu & Ting, 2004; Nosil & Schluter, 2011; Burri et al, 2015), 同时对建立针对濒危物种的保护策略有应用价值(der Sarkissian et al, 2015).基于基因组水平的滑动窗(sliding window)分析可以追溯不同染色体区段的祖先状态, 上面提到过的D-statistics就是一个很好的工具(Kronforst et al, 2013; Smith & Kronforst, 2013). ...
Coalescent-based species tree inference from gene tree topologies under incomplete lineage sorting by maximum likelihood.
Needle morphological evidence of the homoploid hybrid origin of Pinus densata based on analysis of artificial hybrids and the putative parents, Pinus tabuliformis and Pinus yunnanensis.
1
2014
... 大量的调查分析表明, 和生殖隔离密切相关的性状更多地表现出超亲分离, 比如植物的环境适应性和开花物候性状、动物的生活史和行为性状(Rieseberg et al, 1999).总之, 杂种和亲本种间在表型的变异模式上有联系, 但这种联系还没有确定的模式可依.由此可见, 表型在揭示杂交是否存在或者确定亲本来源上有很多限制, 它无法提供确切的依据.但表型指标甚至仅仅是形态指标往往也能提供非常有价值的信息, 比如将它们与基因组水平的标记结合起来可以为物种的界定提供重要信息(Pyron et al, 2016).笔者亲历了一个体现形态数据重要性的实例. 在几十年前对松属(Pinus)的修订中, 我国的分类学家就依据形态特征猜测高山松(Pinus densata)要么来自云南松(P. yunnanensis)和油松(P. tabuliformis)的杂交, 要么作为祖先种分化为另外两种松树(吴中伦, 1956).之后, 研究人员开展了形态、生殖、物候、解剖、幼苗萌发、人工杂交、多重分子标记的群体遗传等多方面的研究, 已有的证据支持了前一个推测(Wang XR et al, 1990, 2001; Wang & Szmidt, 1994; Song et al, 2002, 2003; Ma et al, 2006; Mao et al, 2009; Wang BS et al, 2011; Gao et al, 2012; 张立沙等, 2012; 梁冬等, 2013; Xing et al, 2014; Zhao et al, 2014). ...
Efficient inference of local ancestry.
1
2013
... 已经有一些估算位点祖先状态的计算工具, Padhukasahasram (2014)就进行了较为详尽的综述.一般来说, 这些计算工具需要借助隐马尔科夫模型(hidden Markov models, HMMs)来确定不同亲本来源的基因组区段的界限.HAPMIX软件可以利用未定相(unphased)的基因型数据确定杂种个体特定位点的祖先来源(Price et al, 2009).CRFs应用一个广义隐马尔科夫模型(generalized hidden Markov model), 借助训练集数据实现了对祖先来源的估算(Sankararaman et al, 2012).实现这类分析的软件还包括: SABER (Tang et al, 2006)、HAPA (Sundquist et al, 2008)、LAMP (Sankararaman et al, 2008)、LAMP- LD/LAMP-HAP (Baran et al, 2012)、WINPOP (Pasaniuc et al, 2009)、SupportMix (Omberg et al, 2012)、ASPCA (Moreno-Estrada et al, 2013)、ALLOY (Rodriguez et al, 2013)、RFMix (Maples et al, 2013)、Lanc- CSV (Brown & Pasaniuc, 2014)和EILA (Yang et al, 2013).EILA没有对输入数据位点间连锁平衡的前提假设, 且比LAMP和HAPMIX更准确, 运行速度也不慢.Lanc-CSV实现了经典算法, 但对超大样本数据有速度上的优势. ...
A model-based approach for analysis of spatial structure in genetic data.
1
2012
... 主成分分析(principal component analysis, PCA)是一类经典的多元统计技术, 很早就开始被应用在遗传数据分析中(Menozzi et al, 1978).主成分分析可以将样本间的关系呈现在主成分形成的二维或三维空间上, 但它不能提供杂交存在与否的检验(Patterson et al, 2012), 介于类群间的样本不一定就来自于类群间的杂交(Yang et al, 2012).因此, EIGENSTRAT等软件基于PCA来估算杂交(Price et al, 2006)可能会得出错误的结果.除了EIGENSTRAT, adegenet也是一个主成分分析的工具(Jombart, 2008). ...
Maximum-likelihood models for combined analyses of multiple sequence data.
Coalescent histories on phylogenetic networks and detection of hybridization despite incomplete lineage sorting.
2
2011
... 模型初选后, 应该用恰当的检验来评判选定模型的可靠性.这方面有一系列的工具可以利用, 比如位点内或位点间重组(Mcguire et al, 1997; Kosakovsky et al, 2006; Martin et al, 2015)、各种选择(Delport et al, 2010)、杂交(Yu et al, 2011)、时序性进化速率差异(heterotachy) (Pagel & Meade, 2008)和模型总体上的配合性(Albert & Schluter, 2005; Abby et al, 2012; Ackermann et al, 2016)等.如果初选模型不适合, 那么应该及时调整选用更恰当的模型.如果位点间基因树冲突, 那么可考虑选用应对杂交或者不完全谱系分离的模型, 而不是仍然将序列合并使用, 或者把不相配合的部分数据删除.当然还是要删除那些体现强烈选择印记的、变异饱和的(saturated) (Castresana, 2000; Rodríguezezpeleta et al, 2007; Gnirke et al, 2009)和包含有缺失值(missing)或有缺失变异(deletion)的位点(Lemmon et al, 2009). ...
... 大量的调查分析表明, 和生殖隔离密切相关的性状更多地表现出超亲分离, 比如植物的环境适应性和开花物候性状、动物的生活史和行为性状(Rieseberg et al, 1999).总之, 杂种和亲本种间在表型的变异模式上有联系, 但这种联系还没有确定的模式可依.由此可见, 表型在揭示杂交是否存在或者确定亲本来源上有很多限制, 它无法提供确切的依据.但表型指标甚至仅仅是形态指标往往也能提供非常有价值的信息, 比如将它们与基因组水平的标记结合起来可以为物种的界定提供重要信息(Pyron et al, 2016).笔者亲历了一个体现形态数据重要性的实例. 在几十年前对松属(Pinus)的修订中, 我国的分类学家就依据形态特征猜测高山松(Pinus densata)要么来自云南松(P. yunnanensis)和油松(P. tabuliformis)的杂交, 要么作为祖先种分化为另外两种松树(吴中伦, 1956).之后, 研究人员开展了形态、生殖、物候、解剖、幼苗萌发、人工杂交、多重分子标记的群体遗传等多方面的研究, 已有的证据支持了前一个推测(Wang XR et al, 1990, 2001; Wang & Szmidt, 1994; Song et al, 2002, 2003; Ma et al, 2006; Mao et al, 2009; Wang BS et al, 2011; Gao et al, 2012; 张立沙等, 2012; 梁冬等, 2013; Xing et al, 2014; Zhao et al, 2014). ...
高山松与亲本种多种群在高海拔生境下的苗期适应性研究
1
2012
... 大量的调查分析表明, 和生殖隔离密切相关的性状更多地表现出超亲分离, 比如植物的环境适应性和开花物候性状、动物的生活史和行为性状(Rieseberg et al, 1999).总之, 杂种和亲本种间在表型的变异模式上有联系, 但这种联系还没有确定的模式可依.由此可见, 表型在揭示杂交是否存在或者确定亲本来源上有很多限制, 它无法提供确切的依据.但表型指标甚至仅仅是形态指标往往也能提供非常有价值的信息, 比如将它们与基因组水平的标记结合起来可以为物种的界定提供重要信息(Pyron et al, 2016).笔者亲历了一个体现形态数据重要性的实例. 在几十年前对松属(Pinus)的修订中, 我国的分类学家就依据形态特征猜测高山松(Pinus densata)要么来自云南松(P. yunnanensis)和油松(P. tabuliformis)的杂交, 要么作为祖先种分化为另外两种松树(吴中伦, 1956).之后, 研究人员开展了形态、生殖、物候、解剖、幼苗萌发、人工杂交、多重分子标记的群体遗传等多方面的研究, 已有的证据支持了前一个推测(Wang XR et al, 1990, 2001; Wang & Szmidt, 1994; Song et al, 2002, 2003; Ma et al, 2006; Mao et al, 2009; Wang BS et al, 2011; Gao et al, 2012; 张立沙等, 2012; 梁冬等, 2013; Xing et al, 2014; Zhao et al, 2014). ...
Resolution of deep angiosperm phylogeny using conserved nuclear genes and estimates of early divergence times.
Weak crossability barrier but strong juvenile selection supports ecological speciation of the hybrid pine Pinus densata on the Tibetan Plateau.
1
2014
... 大量的调查分析表明, 和生殖隔离密切相关的性状更多地表现出超亲分离, 比如植物的环境适应性和开花物候性状、动物的生活史和行为性状(Rieseberg et al, 1999).总之, 杂种和亲本种间在表型的变异模式上有联系, 但这种联系还没有确定的模式可依.由此可见, 表型在揭示杂交是否存在或者确定亲本来源上有很多限制, 它无法提供确切的依据.但表型指标甚至仅仅是形态指标往往也能提供非常有价值的信息, 比如将它们与基因组水平的标记结合起来可以为物种的界定提供重要信息(Pyron et al, 2016).笔者亲历了一个体现形态数据重要性的实例. 在几十年前对松属(Pinus)的修订中, 我国的分类学家就依据形态特征猜测高山松(Pinus densata)要么来自云南松(P. yunnanensis)和油松(P. tabuliformis)的杂交, 要么作为祖先种分化为另外两种松树(吴中伦, 1956).之后, 研究人员开展了形态、生殖、物候、解剖、幼苗萌发、人工杂交、多重分子标记的群体遗传等多方面的研究, 已有的证据支持了前一个推测(Wang XR et al, 1990, 2001; Wang & Szmidt, 1994; Song et al, 2002, 2003; Ma et al, 2006; Mao et al, 2009; Wang BS et al, 2011; Gao et al, 2012; 张立沙等, 2012; 梁冬等, 2013; Xing et al, 2014; Zhao et al, 2014). ...
Using shared genomic synteny and shared protein functions to enhance the identification of orthologous gene pairs.
 , 马永鹏
, 马永鹏

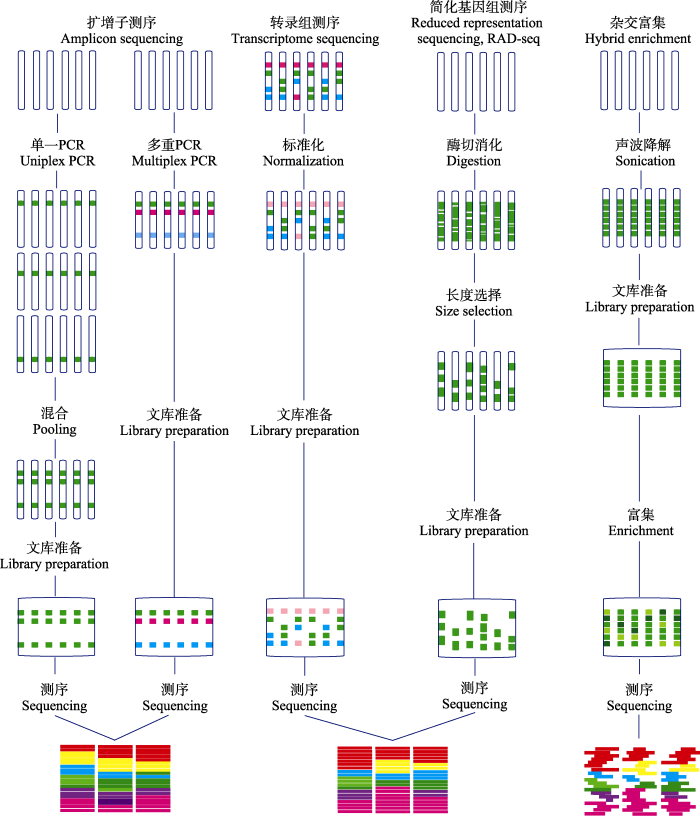
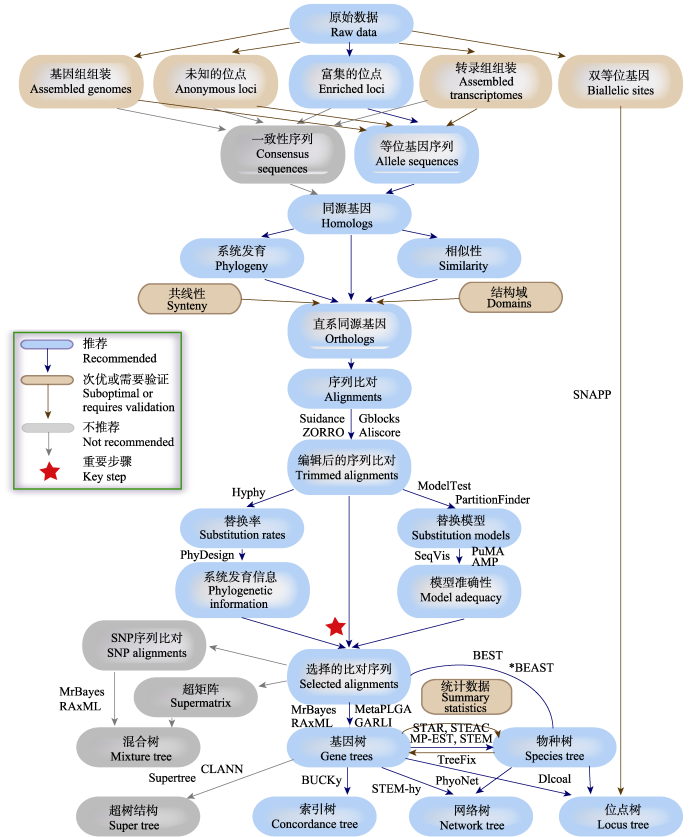
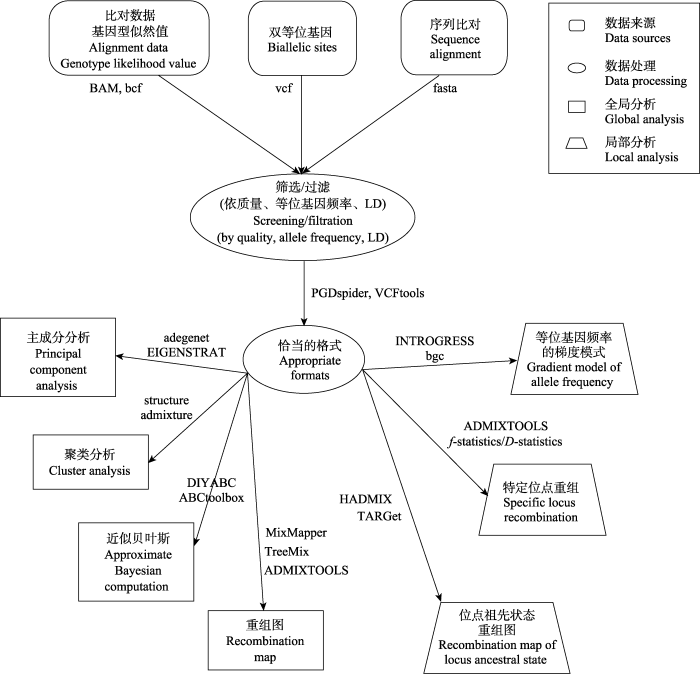
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}