1 Suqian Institute of Agricultural Sciences, Jiangsu Academy of Agricultural Sciences, Suqian, Jiangsu 2238002 College of Life Sciences, Capital Normal University, Beijing 100048
To explore the feasibility of assessing species diversity using DNA barcoding, we investigated this approach by focusing on moths species (Lepidoptera) in Suqian, China. The study evaluated community species richness and rank-abundance curves using the DNA barcoding method, and compared it with the traditional morphology method. Results indicated that there was no significant difference between the DNA barcode-based approach and the morphology-based approach. All DNA barcode-based rank-abundance curves gave similar and clear patterns when compared with morphology-based curves (Kolmogorov-Smirnov two sample test, P > 0.05). Our results indicate that the DNA barcode-based approach is able to be used to estimate species richness.
Keywords:Lepidoptera
;
DNA barcoding
;
species richness
;
rank-abundance curves
QianJin, FenChen, GuijieLuo, WeijiaCai, XuLiu, HaoWang, CaiqingYang, MengdiHao, AibingZhang. Estimation of species richness of moths (Insecta: Lepidoptera) based on DNA barcoding in Suqian, China[J]. Biodiver Sci, 2016, 24(11): 1296-1305 https://doi.org/10.17520/biods.2016202
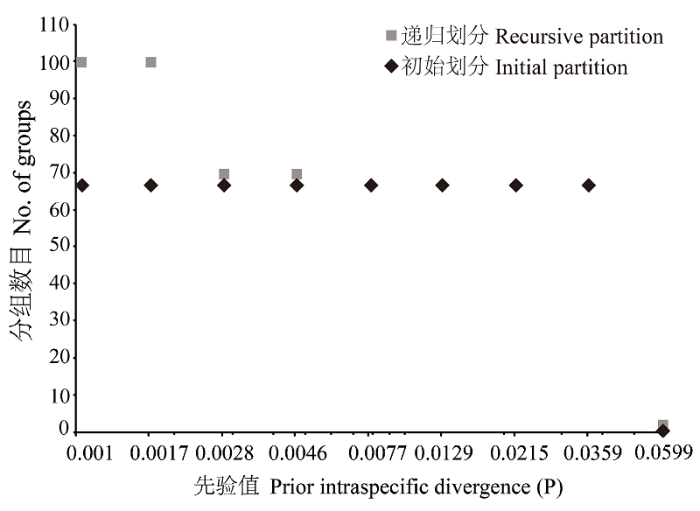
Fig. 1 The automatic partition results of dataset with ABGD method
Table 1
表1
表1 ABGD方法MOTU分组结果与形态种类对照
Table 1 The comparison of ABGD method with morphospecies
MOTU①
种名 Species②
MOTU
种名 Species
MOTU01
Lacanobia contigua
MOTU34
Parapoynx vittalis
MOTU02
Agrotis segetum 黄地老虎
MOTU35
Heliothis assulta 烟青虫
MOTU03
Conogethes punctiferalis 桃蛀螟
MOTU36
Herminia grisealis
MOTU04
Nola cicatricalis
MOTU37
Spodoptera depravata
MOTU05
Hipoepa fractalis 中影单跗夜蛾
MOTU38
Scopula subpunctaria
MOTU06
Oraesia lata 平嘴壶夜蛾
MOTU39
Diaphania indica 瓜绢野螟
MOTU07
Spodoptera litura 斜纹夜蛾
MOTU40
Botyodes diniasalis 黄翅缀叶野螟
MOTU08
Uropyia meticulodina 核桃美舟蛾
MOTU41
Emmelia trabealis 谐夜蛾
MOTU09
Evergestis extimalis
MOTU42
Termioptycha nigrescens
MOTU10
Mythimna separate
MOTU43
Ipimorpha subtusa 杨逸色夜蛾
MOTU11
Ctenoplusia albostriata
MOTU44
Spilosoma lubricipeda
MOTU12
Peridea lativitta 侧带内斑舟蛾
MOTU45
Parapediasia teterrellus
MOTU13
Thyas juno 肖毛翅夜蛾
MOTU46
Adoxophyes orana
MOTU14
Nephopterix fumella
MOTU47
Simplicia rectalis 黑点贫夜蛾
MOTU15
Spodoptera exigua 甜菜夜蛾
MOTU48
Glyptoteles leucacrinella 亮雕斑螟
MOTU16
Oglasa consanguis
MOTU49
Somena scintillans
MOTU17
Diaphania perspectalis 黄杨绢野螟
MOTU50
Anadevidia peponis 葫芦夜蛾
MOTU18
Athetis lepigone
MOTU51
Hypocala subsatura 苹梢鹰夜蛾
MOTU19
Miyakea raddeella
MOTU52
Choristoneura diversana 异色卷蛾
MOTU20
Herpetogramma pseudomagna 狭翅切叶野螟
MOTU53
Choristoneura luticostana 棕色卷蛾
MOTU21
Axylia putris 朽木夜蛾
MOTU54
Archips betulana; Archips podana
MOTU22
Mocis ancilla
MOTU55
Palpita nigropunctalis 白蜡绢须野螟
MOTU23
Mecyna tricolor
MOTU56
Epiblema foenella 白钩小卷蛾
MOTU24
Mamestra brassicae 甘蓝夜蛾
MOTU57
Grammodes geometrica 象夜蛾
MOTU25
Xanthorhoe quadrifasiata
MOTU58; MOTU60
Plusia nadeja
MOTU26
Euproctis similis
MOTU59
Sphinx caligineus
MOTU27
Zanclognatha lunalis 朽镰须夜蛾
MOTU61
Acronicta rumicis 梨剑纹夜蛾
MOTU28
Theretra japonica
MOTU62
Herminia tarsipennalis
MOTU29
Herminia tarsicrinalis
MOTU63
Pangrapta trimantesalis
MOTU30
Eremodrina morosa
MOTU64
Eutelia hamulatrix 沟尾夜蛾
MOTU31
Helicoverpa armigera 棉铃虫
MOTU65
Plusilla rosalia
MOTU32
Nomophila noctuella
MOTU66
Bertula bistrigata
MOTU33
Dysgonia mandschuriana
MOTU67
Gastropacha populifolia 杨枯叶蛾
① Results of ABGD method. ② Results of morphospecies. The grey shaded areas show the difference between ABGD method and morphospecies.①表示基于ABGD方法的MOTU分组结果; ②表示对应的形态物种。ABGD方法与形态种不一致情况以灰色显示。
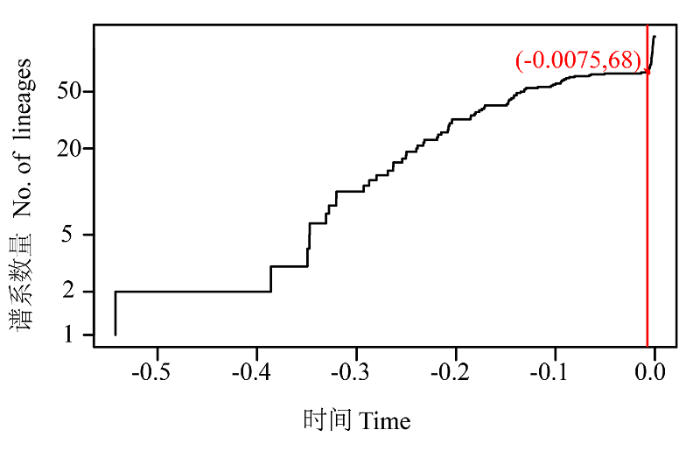
Fig. 2 Lineages-through-time plot based on the time calibrated tree obtained from all 121 haplotypes. The sharp increase in branching rate, corresponding to the transition from interspecies to intraspecies branching events.
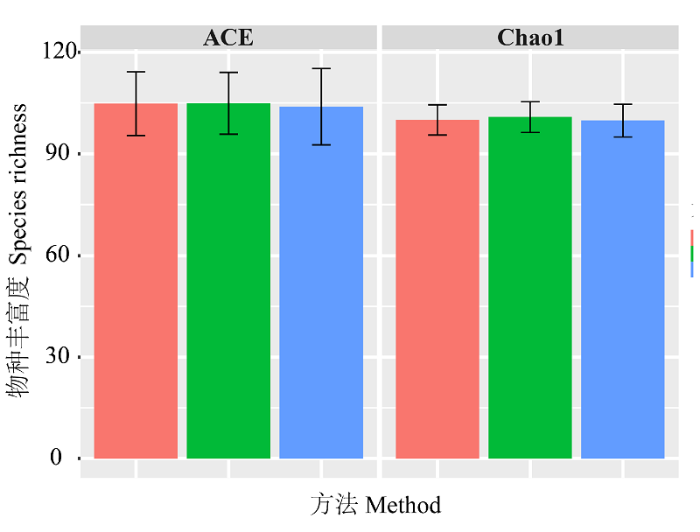
Fig. 3 Species richness estimation results with different methods. Pink colour represents ABGD-based method. Green colour represents GMYC-based method. Blue colour represents morphology-based method. Black error bar indicates confidence interval.
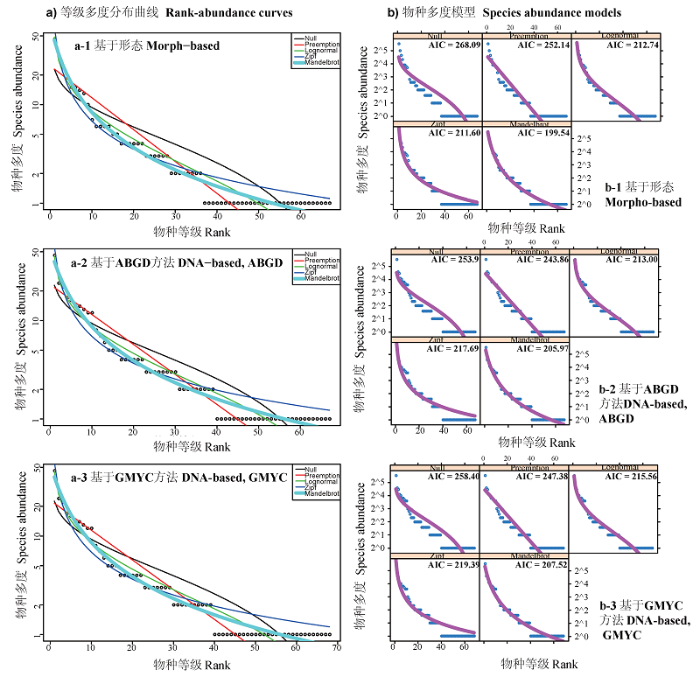
Fig. 4 Rank-abundance curves and fittings of five models. a-1)Species rank-abundance curves based on morphological method, a-2) Species rank-abundance curves based on ABGD method, a-3) Species rank-abundance curves based on GMYC method; b-1) Species abundance models based on morphological method, b-2) Species abundance models based on ABGD method, b-3) Species abundance models based on GMYC method. Black points represent the real abundance data. Black curve represents broken stick model. Red curve represents preemption model. Green curve represents log normal model. Dark blue curve represents Zipf model. Light blue curve represents Zipf-Mandelbrot model.
Appendix 2 MOTU results with GMYC method. The ultrametric tree implemented with BEAST software. Red line indicates transition from between-species to within-species. Red branches represent coalescence processes. Black branches represent speciation and extinction processes. Box shows the MOTU results with GMYC method.
Delimiting species using single-locus data and the Generalized Mixed Yule Coalescent (GMYC) approach: a revised method and evaluation on simulated datasets.
Ten species in one: DNA barcoding reveals cryptic species in the neotropical skipper butterfly Astraptes fulgerator. Proceedings of the National Academy of Sciences,
Species richness and phylogenetic diversity comparisons of soil microbial communities affected by nickel-mining and revegetation efforts in New Caledonia.
A simple 2D non-parametric resampling statistical approach to assess confidence in species identification in DNA barcoding—an alternative to Likelihood and Bayesian approaches.
Mitochondrial phylogenomics and genetic relationships of closely related pine moth (Lasiocampidae: Dendrolimus) species in China, using whole mitochondrial genomes.
Utility of the DNA barcoding gene fragment for parasitic wasp phylogeny (Hymenoptera: Ichneumonoidea): data release and new measure of taxonomic congruence.
... 对于个体小、物种丰富度高的鳞翅目蛾类进行多样性调查的研究中, 通常需要对野外收集到的大量标本进行鉴定, 然后计算各种物种多样性指数.然而, 对于没有分类学背景的生态学者而言, 凭借传统形态学知识对群落内所有物种进行鉴定往往耗时耗力(May, 1988; Blaxter & Floyd, 2003).DNA条形码的兴起打破了这一局面, Hebert等致力于构建一个全球生物多样性数据库(Hebert et al, 2003; Ebach & Holdrege, 2005; Schindel & Miller, 2005; Dincă et al, 2011).截至2016年6月22日, BOLD系统中共记录鳞翅目物种101,790个, 条形码序列1,075,731条(http://www.barcodinglife.org).DNA条形码研究主要分为2种类型: 理论研究和实例研究, 理论研究重点探讨基于DNA条形码的识别方法及有效性, 包括经典的系统发育方法(Saitou & Nei, 1987; Ekrem et al, 2007)、基于聚类的统计学方法(Austerlitz et al, 2009; Jin et al, 2012; 金倩和张爱兵, 2013)、人工智能方法(Zhang et al, 2008, 2012a)以及模糊集合理论(Zhang et al, 2012b); 实例研究是不同生物类群的分类学家通过DNA条形码研究各自感兴趣的生物类群, 例如探讨类群之间的系统发育关系(Qin et al, 2015), 未知物种的分配(Li et al, 2015), 隐存种复合体的研究(Leray & Knowlton, 2015), 对群落内的生物多样性进行测度, 对生态系统的α和β多样性进行估计(Quicke et al, 2012; Jin et al, 2013; Decaëns et al, 2015; Leray & Knowlton, 2015).群落物种丰富度(species richness)、等级多度分布曲线(rank-abundance curves)是测度群落多样性的重要指标, 对群落中种类和数量繁多的鳞翅目昆虫类群来说, 探讨基于分子的方法能否一定程度上代替或补充基于形态的指标体系, 将大大有利于群落多样性的调查和评估. ...
A comprehensive DNA sequence library is essential for identification with DNA barcodes.
1
2007
... 对于个体小、物种丰富度高的鳞翅目蛾类进行多样性调查的研究中, 通常需要对野外收集到的大量标本进行鉴定, 然后计算各种物种多样性指数.然而, 对于没有分类学背景的生态学者而言, 凭借传统形态学知识对群落内所有物种进行鉴定往往耗时耗力(May, 1988; Blaxter & Floyd, 2003).DNA条形码的兴起打破了这一局面, Hebert等致力于构建一个全球生物多样性数据库(Hebert et al, 2003; Ebach & Holdrege, 2005; Schindel & Miller, 2005; Dincă et al, 2011).截至2016年6月22日, BOLD系统中共记录鳞翅目物种101,790个, 条形码序列1,075,731条(http://www.barcodinglife.org).DNA条形码研究主要分为2种类型: 理论研究和实例研究, 理论研究重点探讨基于DNA条形码的识别方法及有效性, 包括经典的系统发育方法(Saitou & Nei, 1987; Ekrem et al, 2007)、基于聚类的统计学方法(Austerlitz et al, 2009; Jin et al, 2012; 金倩和张爱兵, 2013)、人工智能方法(Zhang et al, 2008, 2012a)以及模糊集合理论(Zhang et al, 2012b); 实例研究是不同生物类群的分类学家通过DNA条形码研究各自感兴趣的生物类群, 例如探讨类群之间的系统发育关系(Qin et al, 2015), 未知物种的分配(Li et al, 2015), 隐存种复合体的研究(Leray & Knowlton, 2015), 对群落内的生物多样性进行测度, 对生态系统的α和β多样性进行估计(Quicke et al, 2012; Jin et al, 2013; Decaëns et al, 2015; Leray & Knowlton, 2015).群落物种丰富度(species richness)、等级多度分布曲线(rank-abundance curves)是测度群落多样性的重要指标, 对群落中种类和数量繁多的鳞翅目昆虫类群来说, 探讨基于分子的方法能否一定程度上代替或补充基于形态的指标体系, 将大大有利于群落多样性的调查和评估. ...
Delimiting species using single-locus data and the Generalized Mixed Yule Coalescent (GMYC) approach: a revised method and evaluation on simulated datasets.
Ten species in one: DNA barcoding reveals cryptic species in the neotropical skipper butterfly Astraptes fulgerator. Proceedings of the National Academy of Sciences,
1
2004
... 研究者曾试图建立种内种间遗传距离差异的标准值(标准值为3%或10倍法则)对物种进行阈值划分(Hebert et al, 2004).Smith等(2005)将条形码技术应用于蚂蚁类群的多样性研究中, 并将2-3% COI序列差异作为区分各类群的标准.结果表明, 对于群落中的物种丰富度, 形态学方法和分子可操作单元方法之间并没有显著性的差异.微生物群落研究中已经将3%的16S rRNA阈值作为区分物种的标准(Ovreas, 2000; Oline, 2006; Gomez-Alvarez et al, 2007; Herrera et al, 2007).对于生物多样性调查, 尤其是针对那些没有良好分类学基础的研究而言, 根据DNA条形码阈值法进行物种识别确实行之有效.然而, 该方法不适用于种内种间遗传距离分布存在重叠现象的研究(Hickerson et al, 2006; Rubinoff, 2006; Ward et al, 2009).在本研究中, 我们发现类群之间的平均种间遗传距离约是平均种内遗传距离的8倍, 但是最小种间遗传距离0.0576小于最大种内遗传距离0.1559, 使得种内和种间遗传距离分布出现了重叠(overlap), 种内和种间没有严格定义的DNA条形码空白区(附录3), 如果主观设定一个阈值在本研究中很难奏效. ...
Species richness and phylogenetic diversity comparisons of soil microbial communities affected by nickel-mining and revegetation efforts in New Caledonia.
1
2007
... 研究者曾试图建立种内种间遗传距离差异的标准值(标准值为3%或10倍法则)对物种进行阈值划分(Hebert et al, 2004).Smith等(2005)将条形码技术应用于蚂蚁类群的多样性研究中, 并将2-3% COI序列差异作为区分各类群的标准.结果表明, 对于群落中的物种丰富度, 形态学方法和分子可操作单元方法之间并没有显著性的差异.微生物群落研究中已经将3%的16S rRNA阈值作为区分物种的标准(Ovreas, 2000; Oline, 2006; Gomez-Alvarez et al, 2007; Herrera et al, 2007).对于生物多样性调查, 尤其是针对那些没有良好分类学基础的研究而言, 根据DNA条形码阈值法进行物种识别确实行之有效.然而, 该方法不适用于种内种间遗传距离分布存在重叠现象的研究(Hickerson et al, 2006; Rubinoff, 2006; Ward et al, 2009).在本研究中, 我们发现类群之间的平均种间遗传距离约是平均种内遗传距离的8倍, 但是最小种间遗传距离0.0576小于最大种内遗传距离0.1559, 使得种内和种间遗传距离分布出现了重叠(overlap), 种内和种间没有严格定义的DNA条形码空白区(附录3), 如果主观设定一个阈值在本研究中很难奏效. ...
DNA barcoding will often fail to discover new animal species over broad parameter space.
1
2006
... 研究者曾试图建立种内种间遗传距离差异的标准值(标准值为3%或10倍法则)对物种进行阈值划分(Hebert et al, 2004).Smith等(2005)将条形码技术应用于蚂蚁类群的多样性研究中, 并将2-3% COI序列差异作为区分各类群的标准.结果表明, 对于群落中的物种丰富度, 形态学方法和分子可操作单元方法之间并没有显著性的差异.微生物群落研究中已经将3%的16S rRNA阈值作为区分物种的标准(Ovreas, 2000; Oline, 2006; Gomez-Alvarez et al, 2007; Herrera et al, 2007).对于生物多样性调查, 尤其是针对那些没有良好分类学基础的研究而言, 根据DNA条形码阈值法进行物种识别确实行之有效.然而, 该方法不适用于种内种间遗传距离分布存在重叠现象的研究(Hickerson et al, 2006; Rubinoff, 2006; Ward et al, 2009).在本研究中, 我们发现类群之间的平均种间遗传距离约是平均种内遗传距离的8倍, 但是最小种间遗传距离0.0576小于最大种内遗传距离0.1559, 使得种内和种间遗传距离分布出现了重叠(overlap), 种内和种间没有严格定义的DNA条形码空白区(附录3), 如果主观设定一个阈值在本研究中很难奏效. ...
A simple 2D non-parametric resampling statistical approach to assess confidence in species identification in DNA barcoding—an alternative to Likelihood and Bayesian approaches.
1
2012
... 对于个体小、物种丰富度高的鳞翅目蛾类进行多样性调查的研究中, 通常需要对野外收集到的大量标本进行鉴定, 然后计算各种物种多样性指数.然而, 对于没有分类学背景的生态学者而言, 凭借传统形态学知识对群落内所有物种进行鉴定往往耗时耗力(May, 1988; Blaxter & Floyd, 2003).DNA条形码的兴起打破了这一局面, Hebert等致力于构建一个全球生物多样性数据库(Hebert et al, 2003; Ebach & Holdrege, 2005; Schindel & Miller, 2005; Dincă et al, 2011).截至2016年6月22日, BOLD系统中共记录鳞翅目物种101,790个, 条形码序列1,075,731条(http://www.barcodinglife.org).DNA条形码研究主要分为2种类型: 理论研究和实例研究, 理论研究重点探讨基于DNA条形码的识别方法及有效性, 包括经典的系统发育方法(Saitou & Nei, 1987; Ekrem et al, 2007)、基于聚类的统计学方法(Austerlitz et al, 2009; Jin et al, 2012; 金倩和张爱兵, 2013)、人工智能方法(Zhang et al, 2008, 2012a)以及模糊集合理论(Zhang et al, 2012b); 实例研究是不同生物类群的分类学家通过DNA条形码研究各自感兴趣的生物类群, 例如探讨类群之间的系统发育关系(Qin et al, 2015), 未知物种的分配(Li et al, 2015), 隐存种复合体的研究(Leray & Knowlton, 2015), 对群落内的生物多样性进行测度, 对生态系统的α和β多样性进行估计(Quicke et al, 2012; Jin et al, 2013; Decaëns et al, 2015; Leray & Knowlton, 2015).群落物种丰富度(species richness)、等级多度分布曲线(rank-abundance curves)是测度群落多样性的重要指标, 对群落中种类和数量繁多的鳞翅目昆虫类群来说, 探讨基于分子的方法能否一定程度上代替或补充基于形态的指标体系, 将大大有利于群落多样性的调查和评估. ...
Quantifying species diversity with a DNA barcoding-based method: Tibetan moth species (Noctuidae) on the Qinghai-Tibetan Plateau.
1
2013
... 对于个体小、物种丰富度高的鳞翅目蛾类进行多样性调查的研究中, 通常需要对野外收集到的大量标本进行鉴定, 然后计算各种物种多样性指数.然而, 对于没有分类学背景的生态学者而言, 凭借传统形态学知识对群落内所有物种进行鉴定往往耗时耗力(May, 1988; Blaxter & Floyd, 2003).DNA条形码的兴起打破了这一局面, Hebert等致力于构建一个全球生物多样性数据库(Hebert et al, 2003; Ebach & Holdrege, 2005; Schindel & Miller, 2005; Dincă et al, 2011).截至2016年6月22日, BOLD系统中共记录鳞翅目物种101,790个, 条形码序列1,075,731条(http://www.barcodinglife.org).DNA条形码研究主要分为2种类型: 理论研究和实例研究, 理论研究重点探讨基于DNA条形码的识别方法及有效性, 包括经典的系统发育方法(Saitou & Nei, 1987; Ekrem et al, 2007)、基于聚类的统计学方法(Austerlitz et al, 2009; Jin et al, 2012; 金倩和张爱兵, 2013)、人工智能方法(Zhang et al, 2008, 2012a)以及模糊集合理论(Zhang et al, 2012b); 实例研究是不同生物类群的分类学家通过DNA条形码研究各自感兴趣的生物类群, 例如探讨类群之间的系统发育关系(Qin et al, 2015), 未知物种的分配(Li et al, 2015), 隐存种复合体的研究(Leray & Knowlton, 2015), 对群落内的生物多样性进行测度, 对生态系统的α和β多样性进行估计(Quicke et al, 2012; Jin et al, 2013; Decaëns et al, 2015; Leray & Knowlton, 2015).群落物种丰富度(species richness)、等级多度分布曲线(rank-abundance curves)是测度群落多样性的重要指标, 对群落中种类和数量繁多的鳞翅目昆虫类群来说, 探讨基于分子的方法能否一定程度上代替或补充基于形态的指标体系, 将大大有利于群落多样性的调查和评估. ...
昆虫DNA条形码分析中的距离方法
1
2013
... 对于个体小、物种丰富度高的鳞翅目蛾类进行多样性调查的研究中, 通常需要对野外收集到的大量标本进行鉴定, 然后计算各种物种多样性指数.然而, 对于没有分类学背景的生态学者而言, 凭借传统形态学知识对群落内所有物种进行鉴定往往耗时耗力(May, 1988; Blaxter & Floyd, 2003).DNA条形码的兴起打破了这一局面, Hebert等致力于构建一个全球生物多样性数据库(Hebert et al, 2003; Ebach & Holdrege, 2005; Schindel & Miller, 2005; Dincă et al, 2011).截至2016年6月22日, BOLD系统中共记录鳞翅目物种101,790个, 条形码序列1,075,731条(http://www.barcodinglife.org).DNA条形码研究主要分为2种类型: 理论研究和实例研究, 理论研究重点探讨基于DNA条形码的识别方法及有效性, 包括经典的系统发育方法(Saitou & Nei, 1987; Ekrem et al, 2007)、基于聚类的统计学方法(Austerlitz et al, 2009; Jin et al, 2012; 金倩和张爱兵, 2013)、人工智能方法(Zhang et al, 2008, 2012a)以及模糊集合理论(Zhang et al, 2012b); 实例研究是不同生物类群的分类学家通过DNA条形码研究各自感兴趣的生物类群, 例如探讨类群之间的系统发育关系(Qin et al, 2015), 未知物种的分配(Li et al, 2015), 隐存种复合体的研究(Leray & Knowlton, 2015), 对群落内的生物多样性进行测度, 对生态系统的α和β多样性进行估计(Quicke et al, 2012; Jin et al, 2013; Decaëns et al, 2015; Leray & Knowlton, 2015).群落物种丰富度(species richness)、等级多度分布曲线(rank-abundance curves)是测度群落多样性的重要指标, 对群落中种类和数量繁多的鳞翅目昆虫类群来说, 探讨基于分子的方法能否一定程度上代替或补充基于形态的指标体系, 将大大有利于群落多样性的调查和评估. ...
昆虫DNA条形码分析中的距离方法
1
2013
... 对于个体小、物种丰富度高的鳞翅目蛾类进行多样性调查的研究中, 通常需要对野外收集到的大量标本进行鉴定, 然后计算各种物种多样性指数.然而, 对于没有分类学背景的生态学者而言, 凭借传统形态学知识对群落内所有物种进行鉴定往往耗时耗力(May, 1988; Blaxter & Floyd, 2003).DNA条形码的兴起打破了这一局面, Hebert等致力于构建一个全球生物多样性数据库(Hebert et al, 2003; Ebach & Holdrege, 2005; Schindel & Miller, 2005; Dincă et al, 2011).截至2016年6月22日, BOLD系统中共记录鳞翅目物种101,790个, 条形码序列1,075,731条(http://www.barcodinglife.org).DNA条形码研究主要分为2种类型: 理论研究和实例研究, 理论研究重点探讨基于DNA条形码的识别方法及有效性, 包括经典的系统发育方法(Saitou & Nei, 1987; Ekrem et al, 2007)、基于聚类的统计学方法(Austerlitz et al, 2009; Jin et al, 2012; 金倩和张爱兵, 2013)、人工智能方法(Zhang et al, 2008, 2012a)以及模糊集合理论(Zhang et al, 2012b); 实例研究是不同生物类群的分类学家通过DNA条形码研究各自感兴趣的生物类群, 例如探讨类群之间的系统发育关系(Qin et al, 2015), 未知物种的分配(Li et al, 2015), 隐存种复合体的研究(Leray & Knowlton, 2015), 对群落内的生物多样性进行测度, 对生态系统的α和β多样性进行估计(Quicke et al, 2012; Jin et al, 2013; Decaëns et al, 2015; Leray & Knowlton, 2015).群落物种丰富度(species richness)、等级多度分布曲线(rank-abundance curves)是测度群落多样性的重要指标, 对群落中种类和数量繁多的鳞翅目昆虫类群来说, 探讨基于分子的方法能否一定程度上代替或补充基于形态的指标体系, 将大大有利于群落多样性的调查和评估. ...
Log-series and log-normal parameters as diversity discriminants for the Lepidoptera.
Mitochondrial phylogenomics and genetic relationships of closely related pine moth (Lasiocampidae: Dendrolimus) species in China, using whole mitochondrial genomes.
1
2015
... 对于个体小、物种丰富度高的鳞翅目蛾类进行多样性调查的研究中, 通常需要对野外收集到的大量标本进行鉴定, 然后计算各种物种多样性指数.然而, 对于没有分类学背景的生态学者而言, 凭借传统形态学知识对群落内所有物种进行鉴定往往耗时耗力(May, 1988; Blaxter & Floyd, 2003).DNA条形码的兴起打破了这一局面, Hebert等致力于构建一个全球生物多样性数据库(Hebert et al, 2003; Ebach & Holdrege, 2005; Schindel & Miller, 2005; Dincă et al, 2011).截至2016年6月22日, BOLD系统中共记录鳞翅目物种101,790个, 条形码序列1,075,731条(http://www.barcodinglife.org).DNA条形码研究主要分为2种类型: 理论研究和实例研究, 理论研究重点探讨基于DNA条形码的识别方法及有效性, 包括经典的系统发育方法(Saitou & Nei, 1987; Ekrem et al, 2007)、基于聚类的统计学方法(Austerlitz et al, 2009; Jin et al, 2012; 金倩和张爱兵, 2013)、人工智能方法(Zhang et al, 2008, 2012a)以及模糊集合理论(Zhang et al, 2012b); 实例研究是不同生物类群的分类学家通过DNA条形码研究各自感兴趣的生物类群, 例如探讨类群之间的系统发育关系(Qin et al, 2015), 未知物种的分配(Li et al, 2015), 隐存种复合体的研究(Leray & Knowlton, 2015), 对群落内的生物多样性进行测度, 对生态系统的α和β多样性进行估计(Quicke et al, 2012; Jin et al, 2013; Decaëns et al, 2015; Leray & Knowlton, 2015).群落物种丰富度(species richness)、等级多度分布曲线(rank-abundance curves)是测度群落多样性的重要指标, 对群落中种类和数量繁多的鳞翅目昆虫类群来说, 探讨基于分子的方法能否一定程度上代替或补充基于形态的指标体系, 将大大有利于群落多样性的调查和评估. ...
Utility of the DNA barcoding gene fragment for parasitic wasp phylogeny (Hymenoptera: Ichneumonoidea): data release and new measure of taxonomic congruence.
1
2012
... 对于个体小、物种丰富度高的鳞翅目蛾类进行多样性调查的研究中, 通常需要对野外收集到的大量标本进行鉴定, 然后计算各种物种多样性指数.然而, 对于没有分类学背景的生态学者而言, 凭借传统形态学知识对群落内所有物种进行鉴定往往耗时耗力(May, 1988; Blaxter & Floyd, 2003).DNA条形码的兴起打破了这一局面, Hebert等致力于构建一个全球生物多样性数据库(Hebert et al, 2003; Ebach & Holdrege, 2005; Schindel & Miller, 2005; Dincă et al, 2011).截至2016年6月22日, BOLD系统中共记录鳞翅目物种101,790个, 条形码序列1,075,731条(http://www.barcodinglife.org).DNA条形码研究主要分为2种类型: 理论研究和实例研究, 理论研究重点探讨基于DNA条形码的识别方法及有效性, 包括经典的系统发育方法(Saitou & Nei, 1987; Ekrem et al, 2007)、基于聚类的统计学方法(Austerlitz et al, 2009; Jin et al, 2012; 金倩和张爱兵, 2013)、人工智能方法(Zhang et al, 2008, 2012a)以及模糊集合理论(Zhang et al, 2012b); 实例研究是不同生物类群的分类学家通过DNA条形码研究各自感兴趣的生物类群, 例如探讨类群之间的系统发育关系(Qin et al, 2015), 未知物种的分配(Li et al, 2015), 隐存种复合体的研究(Leray & Knowlton, 2015), 对群落内的生物多样性进行测度, 对生态系统的α和β多样性进行估计(Quicke et al, 2012; Jin et al, 2013; Decaëns et al, 2015; Leray & Knowlton, 2015).群落物种丰富度(species richness)、等级多度分布曲线(rank-abundance curves)是测度群落多样性的重要指标, 对群落中种类和数量繁多的鳞翅目昆虫类群来说, 探讨基于分子的方法能否一定程度上代替或补充基于形态的指标体系, 将大大有利于群落多样性的调查和评估. ...

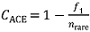
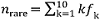
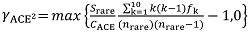


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}