芸薹属植物全基因组重测序研究进展
罗瑞, 陈娅, 张汉马
生物多样性
2023, 31 ( 10):
23237-.
DOI: 10.17520/biods.2023237
芸薹属(Brassica)包含多种重要经济作物。近年来, 随着DNA测序技术的快速发展和测序成本的不断降低, 全基因组重测序已成为芸薹属植物研究的重要手段。本文简要介绍了芸薹属植物全基因组测序近况以及在利用重测序技术研究该属植物的驯化起源、环境适应性和重要性状形成的分子基础等方面所取得的一些重要进展, 包括目前已经全测序的8个芸苔属植物基因组, 利用全基因组重测序技术研究甘蓝、白菜、芥菜等物种的驯化起源及关系, 鉴定芸苔属植物中与病害、节律、矿质养分响应以及与色素合成、雄性不育、重要生产性状相关基因等方面取得的重要进展。这些进展不仅展示了全基因组重测序技术与前期基于线粒体DNA和叶绿体DNA标记技术相比在研究芸苔属植物的优势, 也为培育具有优良环境适应性和优良生产性状的芸苔属作物品种提供了重要理论和技术支撑。未来, 全基因组重测序技术在芸薹属植物研究中将会进一步普及, 其对芸苔属植物的基础生物学研究和新品种培育的影响将会得到彰显。
View image in article
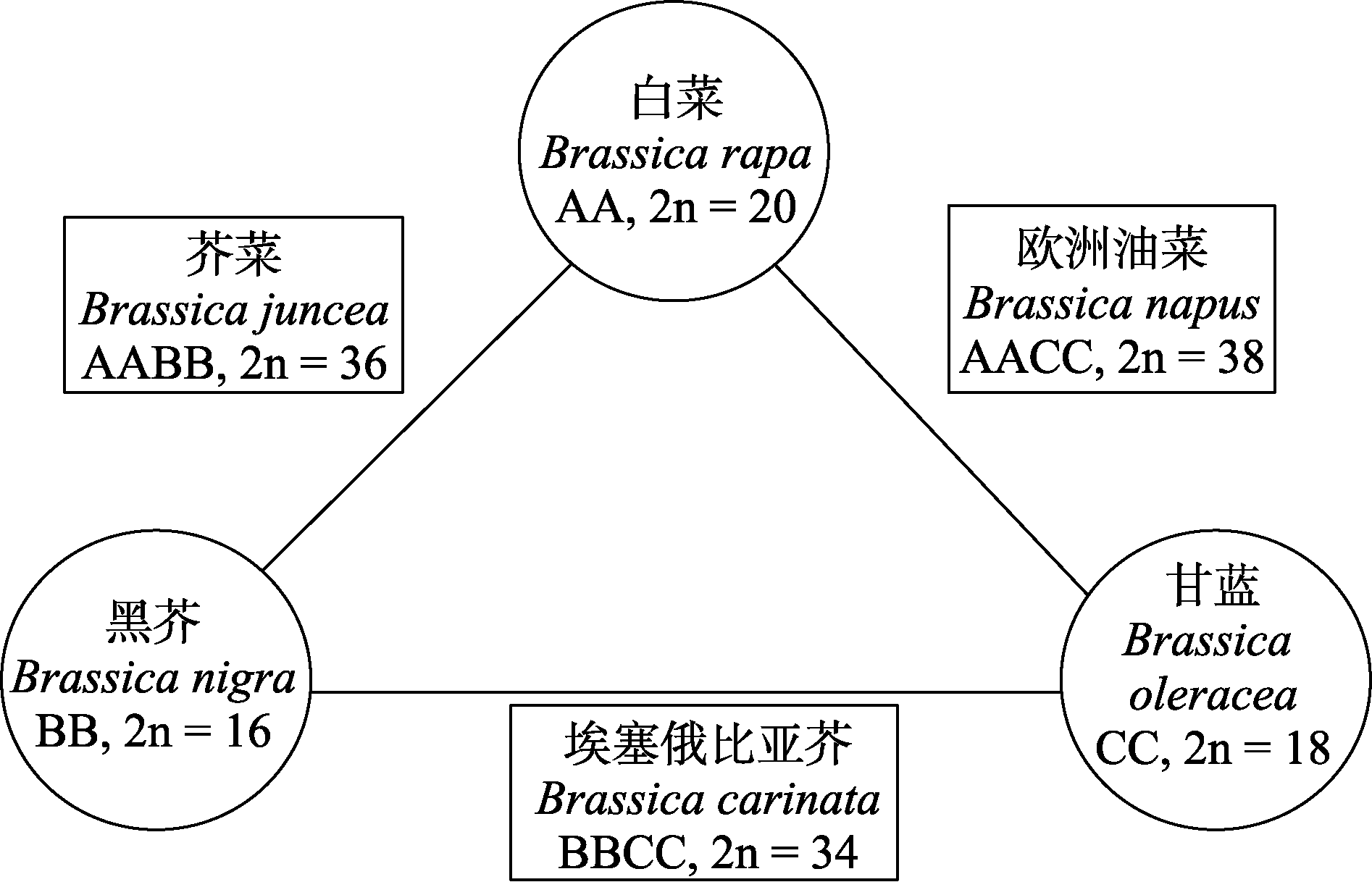
图1
芸薹属禹氏三角理论
正文中引用本图/表的段落
芸薹属(Brassica)是十字花科中最具经济价值的一个属, 包含多种重要的蔬菜、油料和饲用作物(Williams & Hill, 1986; 周灿等, 2014)。该属植物起源于西欧、环地中海地区及亚洲的温带地区, 广泛分布于世界各地。全球有38个种, 中国大约有15个种(Cheng et al, 2013)。禹氏三角理论(图1)将芸薹属植物分为3个基本的二倍体种, 即白菜(Brassica rapa, AA, 2n = 20)、黑芥(B. nigra, BB, 2n = 16)、甘蓝(B. oleracea, CC, 2n = 18)和3个四倍体杂交复合种, 即欧洲油菜(甘蓝型油菜) (B. napus, AACC, 2n = 38)、芥菜(B. juncea, AABB, 2n = 36)、埃塞俄比亚芥(B. carinata, BBCC, 2n = 34) (Nagaharu & Nagaharu, 1935)。
芸薹属植物由于长期自然选择与人工驯化选择的影响, 衍生了多个亚种、变种及不同的生态型, 从而使研究芸薹属植物驯化起源成为植物学研究的热点之一.在全基因组测序之前, 研究者主要利用线粒体DNA、叶绿体DNA和微卫星标记等开展芸薹属驯化起源方面的研究, 这些研究方法受限制于DNA序列长度的影响而具有相当大的局限性(Palmer et al, 1983; Qiao et al, 2016; Thakur et al, 2018; You et al, 2022).随着全基因组测序和重测序数据的累积, 为利用分子生物学手段研究芸薹属驯化起源提供了更加充足的数据, 弥补了线粒体DNA和叶绿体DNA等早期分子标记方法的不足.Cheng等(2016)利用199份白菜和119份甘蓝不同类型材料的重测序数据, 区分出6个白菜群体和7个甘蓝群体.基于系统发育树发现芜菁(B. rapa subsp. rapa)和沙菜籽(B. rapa var. sarson)组成的进化支最接近白菜发育树的根, 表明这两个群体是最后一个白菜共同祖先的早期分化, 而大白菜(B. rapa subsp. pekinensis)则处于根最远的进化支; 大头菜(B. oleracea var. gongylodes)和野生甘蓝(wild B. oleracea)进化支与甘蓝的根较近, 卷心菜(B. oleracea var. capitata)处于最远的进化支.有趣的是, 研究发现大白菜与卷心菜具有相似的叶片形态, 表明这两个群体从祖先分化出来之后独立发展成相似的形态, 这为理解趋同亚基因组的平行选择提供了参考.Qi等(2017)基于全球126份白菜获得超过31,000个全基因组SNPs, 揭示了5个不同遗传群体的起源证据.研究认为南亚和东亚类群的小白菜(B. rapa subsp. chinensis)、大白菜和印度菜籽(B. rapa subsp. trilocularis)可能是单系类群; 油菜花和印度油菜可能是多系起源.同时, 研究也表示没有证据显示西兰花拉贝是野生型和最早驯化的白菜亚种.种群历史动态分析揭示了白菜在2,400-4,100年前传入亚洲, 大白菜是在1,200-2,100年前由小白菜和欧洲-中亚的白菜混合而成.Lu等(2019)构建了来自全世界的3种生态型(春季、冬季和半冬季)的588份甘蓝型油菜的基因组变异数据, 推测出甘蓝型油菜是7,290年前由驯化的油菜与甘蓝的杂交而来, 从基因组水平上验证了此前通过Ks估算的起源时间(Chalhoub et al, 2014; Sun et al, 2017).同时, 该研究分别探讨了甘蓝型油菜的A亚基因组和C亚基因组的不同进化来源, 即A亚基因组是由欧洲大头菜(B. rapa subsp. rapa)的祖先进化而来, 而C亚基因组是由甘蓝、花椰菜(B. oleracea var. botrytis)、西兰花(B. oleracea var. italica)和芥蓝(B. oleracea var. alboglabra)的共同祖先进化而来, 为甘蓝型油菜的各亚基因组的来源与进化过程提供了新证据.通过连锁不平衡和种群历史动态分析揭示了冬季油菜为甘蓝型油菜的原始种群, 春季、半冬季和非油籽甘蓝型油菜分别形成于416、60和277年前, 这些结果与史实记载相符.总而言之, 该研究从全基因组水平上解析了甘蓝型油菜的起源和驯化改良历史.Yang等(2016)利用18份芥菜、5份甘蓝型油菜和27份白菜的重测序数据发现芥菜A亚基因组和甘蓝型油菜A亚基因组具有独立的地理起源, 即芥菜A亚基因组来源于亚洲的白菜, 而甘蓝型油菜来源于欧洲萝卜.此外, 研究还表明所有的芥菜品系的A亚基因组属于单系起源, 而后根据用途进化形成不同的亚种.同时, 通过系统发育分析和贝叶斯方法推断芥菜大约形成于39,000-55,000年前, 有效解决了同义核苷酸取代率估计新多倍体物种形成时间不准确的问题.Kang等(2021)基于PacBio长片段测序组装的黄籽芥菜(B. juncea)基因组图谱和480份重测序材料提出芥菜单系起源于8,000-14,000年前的西亚, 并推测出过去500-5,000年里至少存在3次独立的驯化事件, 包括中亚附近的籽芥、印度次大陆的油籽芥和东亚的根芥.在这3次独立的驯化后产生了中国西北部的黄籽芥菜、四川盆地的茎芥和印度东部的阔叶芥菜等.该研究阐明了芥菜的复杂的进化和驯化历史.上述研究显示, 全基因组重测序为研究芸薹属植物的起源进化和驯化改良历史提供了新的依据. ... Fine mapping and whole-genome resequencing identify the seed coat color gene in Brassica rapa 1 2016 ... 在其他方面, 利用全基因组重测序对大黄油菜(B. rapa var. oleifera)和09A-126两种白菜系构建的近等基因系BC4群体分析筛选到影响种皮颜色的候选基因BrTT1 (Wang YH et al, 2016).通过全基因组重测序比较野生型与经甲烷磺酸乙酯诱变的大白菜定位到控制白菜淡绿色的基因BrCAO, 序列分析表明BrCAO第一个内含子的核苷酸突变导致蛋白质翻译被终止, 这为探索BrCAO的功能和获得淡绿色大白菜提供了有效途径(Zhao YH et al, 2022).研究者通过分析芥菜系紫叶突变体获得了控制紫叶性状与花青素合成的候选基因BjPI1, 并围绕该基因获得了10个AFLP标记, 为紫叶基因的克隆、芸薹属植物的选育和阐明花青素积累的分子机制奠定基础(Zhao et al, 2017).利用全基因组重测序对193份云贵高原芥菜分析获得211个与种皮颜色相关的基因, 包括15个已知参与类黄酮生物合成途径和2个(BjuB018407和BjuA034936)与芥菜黄籽显著相关的基因.有趣的是, 该研究发现这些与种皮颜色相关的基因在A亚基因组和B亚基因组的分布并不均匀, 但受限于目前芥菜B亚基因组的研究较少, 而未获得更多的信息.因此, 推动芥菜B亚基因组的研究将对进一步改善种皮性状具有重要意义(Yuan et al, 2023). ... WHITE PANICLE1, a val-tRNA synthetase regulating chloroplast ribosome biogenesis in rice, is essential for early chloroplast development 1 2016 ... 色素不仅在植物的体色和花色的表型中起着重要作用, 也对植物生长发育至关重要.其主要包括脂溶性的叶绿体色素和水溶性的细胞液色素.叶绿素是植物光合器官和色素的重要组成部分, 缺乏叶绿素会破坏植物代谢, 进而对植物产出带来影响(Czarnecki & Grimm, 2012).如甘蓝型油菜BnaA03.CHLH编码序列的SNPs可引起CHLH活性降低, 并抑制转录光系统I、II的调控基因的RNA聚合酶活性, 导致叶绿素a/b和类胡萝卜素含量降低, 进而引起叶绿体超微结构退化(Zhao et al, 2020).这为解析叶绿素生物合成和叶绿体发育的复杂多基因调控途径提供参考.多项研究表明, 即使是点突变也会导致整个调控网络的变化(Wang YL et al, 2016; Ge et al, 2017).因此, 我们相信由全基因组重测序获得的大量SNPs将为解析这一复杂调控途径提供重要保障. ... Genetic and molecular analysis reveals that two major loci and their interaction confer clubroot resistance in canola introgressed from rutabaga 1 2022 ... 生物病害是常见的生物胁迫因素, 常常使植物的正常生长发育受到干扰和破坏, 从而导致产量降低、品质变劣, 甚至局部或全株死亡.据不完全统计, 全世界粮食因病害而减产10%、蔬菜减产达40%.为此, 众多研究者利用全基因组重测序技术挖掘了大量芸薹属植物病害适应性基因, 为定向选育高抗病株系以实现作物增产增收提供了充足的理论依据.基于全基因组重测序技术, Tollenaere等(2012)对甘蓝型油菜两个抗Rlm4的强候选群体进行分析, 挖掘到18个抗黑胫病候选基因BLR1-BLR18; Inturrisi等(2018)获取了11份芥菜的高质量SNPs, 挖掘到抗白锈病基因AcB1-A4.1和AcB1-A5.1以及抗黑腿基因LMJR1、PhR2和LMJR2; Lee等(2015)以卷心菜自交系C1184 (黑腐病敏感)和C1234 (黑腐病抗性)为亲本建立定位群体, 鉴定到21个抗黑腐病潜在抗性基因和4个可能增加抗性的数量性状基因位点 (quantitative trait locus, QTL)区域; Liu等(2017)对通过小孢子培养技术从01-20 (高易感)和卷心菜96-100 (高抗性)的杂交中培养出230个双单倍体系卷心菜进行分析, 挖掘到24个In/Del标记和与枯萎病抗性相关基因FOC1.虽然在这里仅介绍了部分病害挖掘工作, 但我们可利用全基因组重测序技术转移到其他任何性状的研究, 如根肿病等(Kopec et al, 2021; Wang et al, 2022).根据这些全基因重测序的结果, 可利用抗病特异性标记等实验方法建立快速培育和筛选抗病株系的技术体系, 高效完成定向选育高抗病株系的工作, 从而协助植物应对生物病害.值得注意的是, 有研究者利用88株芥菜-灌木芸薹(B. fruticulosa)侵染系(introgression lines, ILs)植株进行基因分型测序, 在7条芥菜染色体上发现了20个基因可能参与对茎腐病的抗性作用, 这为将外来抗性位点(introgressed resistant loci)转移到芸薹属的农艺优势品种中提供了重要的参考价值(Atri et al, 2019), 也为解决抗性复杂遗传基础给传统育种带来的挑战提供了更多的契机(Wei et al, 2017). ... Construction of restorer lines and molecular mapping for restorer gene of hau cytoplasmic male sterility in Brassica napus 1 2019 ... 雄性不育是芸薹属作物中产生杂种优势的重要原因(许忠民等, 2007), 挖掘雄性不育性状相关的基因不仅对解析其遗传机制提供分子依据, 也可为芸薹属杂交育种提供重要的技术支撑.利用全基因组重测序分别在甘蓝型油菜突变体和大白菜突变体中鉴定到参与雄性和雌性不育调控的重要基因BnaC03g56870D (Teng et al, 2017)和Bra010198 (Tan et al, 2019), 为解析不同芸薹属植物雄性不育的遗传机制提供了重要依据.与此同时, 大白菜细胞质雄性不育(cytoplasmic male sterility, CMS)恢复系的In/Del标记SC718的发现, 为分子标记辅助育种培育和筛选新细胞质雄性不育恢复系提供了参考(Zhang et al, 2017).根据雄性不育和雄性不育恢复的关联, 利用细胞质雄性不育的甘蓝型油菜(B. napus)恢复系又解析了恢复基因Rfh, 为研究恢复基因和CMS基因之间的协同进化以及通过分子育种培育优良杂种提供了新的依据(Wei et al, 2019).此外, 研究者基于5个育种恢复系芥菜的全基因组重测序数据开发了KASPar标记, 用于辅助标记Rfo的转移和生产批次杂交水平测试(Gudi et al, 2020). ... Genome-wide association mapping of resistance to a Brazilian isolate of Sclerotinia sclerotiorum in soybean genotypes mostly from Brazil 1 2017 ... 生物病害是常见的生物胁迫因素, 常常使植物的正常生长发育受到干扰和破坏, 从而导致产量降低、品质变劣, 甚至局部或全株死亡.据不完全统计, 全世界粮食因病害而减产10%、蔬菜减产达40%.为此, 众多研究者利用全基因组重测序技术挖掘了大量芸薹属植物病害适应性基因, 为定向选育高抗病株系以实现作物增产增收提供了充足的理论依据.基于全基因组重测序技术, Tollenaere等(2012)对甘蓝型油菜两个抗Rlm4的强候选群体进行分析, 挖掘到18个抗黑胫病候选基因BLR1-BLR18; Inturrisi等(2018)获取了11份芥菜的高质量SNPs, 挖掘到抗白锈病基因AcB1-A4.1和AcB1-A5.1以及抗黑腿基因LMJR1、PhR2和LMJR2; Lee等(2015)以卷心菜自交系C1184 (黑腐病敏感)和C1234 (黑腐病抗性)为亲本建立定位群体, 鉴定到21个抗黑腐病潜在抗性基因和4个可能增加抗性的数量性状基因位点 (quantitative trait locus, QTL)区域; Liu等(2017)对通过小孢子培养技术从01-20 (高易感)和卷心菜96-100 (高抗性)的杂交中培养出230个双单倍体系卷心菜进行分析, 挖掘到24个In/Del标记和与枯萎病抗性相关基因FOC1.虽然在这里仅介绍了部分病害挖掘工作, 但我们可利用全基因组重测序技术转移到其他任何性状的研究, 如根肿病等(Kopec et al, 2021; Wang et al, 2022).根据这些全基因重测序的结果, 可利用抗病特异性标记等实验方法建立快速培育和筛选抗病株系的技术体系, 高效完成定向选育高抗病株系的工作, 从而协助植物应对生物病害.值得注意的是, 有研究者利用88株芥菜-灌木芸薹(B. fruticulosa)侵染系(introgression lines, ILs)植株进行基因分型测序, 在7条芥菜染色体上发现了20个基因可能参与对茎腐病的抗性作用, 这为将外来抗性位点(introgressed resistant loci)转移到芸薹属的农艺优势品种中提供了重要的参考价值(Atri et al, 2019), 也为解决抗性复杂遗传基础给传统育种带来的挑战提供了更多的契机(Wei et al, 2017). ... Inheritance and genetic mapping of late-bolting to early-bolting gene, BrEb-1, in Chinese cabbage (Brassica rapa L.) 1 2022 ... 此外, 研究者也对另一生物节律行为——抽薹开展了相关研究.植物的抽薹行为主要发生在花芽分化后, 也受开花调控的影响.通过重测序技术在两个大白菜早抽苔突变体ebm5-1和ebm5-2中鉴定到BrSDG8基因与大白菜抽薹相关, 并明确了该基因编码H3K4me3的蛋白质(Fu et al, 2020), 与H3K36me3共同参与拟南芥的FLC表达的激活(Pien et al, 2008; Xu et al, 2008).Wei等(2022)也成功挖掘出大白菜A07染色体上的3个延迟抽薹性状的潜在候选基因BraA07g029500、BraA07g029530和BraA07g030360.研究人员利用109个芥菜自然群体材料重测序数据揭示了CK2B1在芥菜茎膨大形成中的作用, 并进一步表明CK2B1与细胞周期G1/S转变因子E2Fa的相互作用, 阐明了控制芥菜茎膨大形成的机制, 为提高芥菜营养器官形成的产量提供了有效的分子靶点(Zhang et al, 2020). ... Ecological genomics and process modeling of local adaptation to climate 1 2014 ... 芸薹属作物的栽培遍布世界各地, 它们如何适应不同气候、环境以及各种各样的病虫害等是植物学研究中的重要课题(Skelly et al, 2007; Balanyà et al, 2009).全基因组测序和重测序数据为研究芸薹属作物环境适应的分子机理提供了新的可能性(Savolainen et al, 2013; Weinig et al, 2014).在下文中, 我们将简要介绍全基因组测序和重测序数据在研究芸薹属作物在病害、节律和矿物元素等适应性方面所取得的部分进展. ... Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome 1 2019 ... 全基因组重测序(whole-genome resequencing)通过对已有参考基因组序列的物种进行个体或群体全基因组测序, 从而检测出全基因组范围的单核苷酸多态性(single nucleotide polymorphism, SNP)、基因组结构变异(structure variation, SV)、插入缺失变异位点(insertion/deletion, In/Del)、拷贝数变异(copy number variation, CNV)等变异信息, 获得个体或群体分子遗传特征, 并进行重要经济性状基因筛选预测及遗传进化分析(李国治和邓卫东, 2018).自1977年利用第一代Sanger测序法(双脱氧末端终止法)绘制完整的基因图谱以来(Sanger et al, 1977), 目前已发展到第三代测序技术.第二代测序技术因具有高通量、低成本和高准确性等特点, 是目前全基因组重测序的主要测序手段, 主要包括454测序技术(Margulies et al, 2005)、Illumina/Solexa测序技术(Seo et al, 2005)和SOLiD测序技术(Mardis, 2008).454测序技术利用焦磷酸法测序, 使用串联平行合成原理, 通过将DNA片段连接在微球上, 同时在DNA合成过程中检测释放的光信号, 从而实现高通量的DNA测序; 其测序读长通常在400-700 bp之间, 测序误差率通常在1%-2%左右.Illumina/Solexa测序技术利用桥式PCR和碱基扩增原理, 通过反复周期性地加入荧光标记的核苷酸并检测释放的光信号, 实现高通量、高精度的DNA测序; 其测序读长通常可达到数百个碱基对, 测序通量是第一代测序技术的数千倍, 而测序错误率通常在0.1%-1%之间.SOLiD测序技术采用特殊的二碱基编码技术, 通过短DNA片段的反复循环合成和测序来实现高通量的DNA测序; 其测序读长通常为50-75个碱基对, 错误率通常在1%-2%左右.此外, 以Heliscope单分子技术、SMRT技术和纳米孔单分子技术为代表的第三代测序技术也逐步应用于科学研究.Heliscope技术和SMRT技术是利用荧光信号进行测序, 而纳米孔单分子技术则是利用不同碱基产生的电信号进行测序.这些三代测序技术的测序读长通常在数千个碱基对到数十万个碱基对之间, 但错误率通常在5%-15%之间.虽然第三代测序技术的长读长为研究提供了许多优势, 但其较低的精确度可能需要在数据分析中采取更加谨慎的措施, 以确保得到准确的测序结果(闫绍鹏等, 2012).随着测序技术的发展和改进, 高保真测序技术为科学研究带来了更多的机会.高保真测序技术利用荧光信号进行测序, 并通过多次循环合成和错误校正序列获取; 其测序读长可达数万个碱基对以上, 并且准确度可达99%以上(Wenger et al, 2019).这为解决基因组组装、基因变异检测等科学问题提供了更便捷的技术平台. ... Rapid-cycling populations of Brassica 1 1986 ... 芸薹属(Brassica)是十字花科中最具经济价值的一个属, 包含多种重要的蔬菜、油料和饲用作物(Williams & Hill, 1986; 周灿等, 2014).该属植物起源于西欧、环地中海地区及亚洲的温带地区, 广泛分布于世界各地.全球有38个种, 中国大约有15个种(Cheng et al, 2013).禹氏三角理论(图1)将芸薹属植物分为3个基本的二倍体种, 即白菜(Brassica rapa, AA, 2n = 20)、黑芥(B. nigra, BB, 2n = 16)、甘蓝(B. oleracea, CC, 2n = 18)和3个四倍体杂交复合种, 即欧洲油菜(甘蓝型油菜) (B. napus, AACC, 2n = 38)、芥菜(B. juncea, AABB, 2n = 36)、埃塞俄比亚芥(B. carinata, BBCC, 2n = 34) (Nagaharu & Nagaharu, 1935). ... Whole-genome resequencing of a worldwide collection of rapeseed accessions reveals the genetic basis of ecotype divergence 1 2019 ... 生物节律是生物在起源和进化早期对环境中光、地磁等地球物理因素节律性变化的适应现象, 对植物的生命周期、产量和种子的品质起着至关重要的作用, 是目前植物学领域的一个新的研究热点(门中华和李生秀, 2009).研究表明, 模式植物拟南芥的开花位点T (FT)调控着与响应环境适应和发育信号相关的多种开花途径(Srikanth & Schmid, 2011; Andrés & Coupland, 2012), 但受到开花位点C (FLC)的抑制(Searle et al, 2006).使用来自全球991份甘蓝型油菜全基因组重测序数据集也证实了在甘蓝型油菜相同表达模式, 3种甘蓝型油菜生态型对不同地理环境条件的适应能力因FT与FLC的上游50 bp的特异性SNP而发生改变, 这一变化也获得了转录组学数据的支持, 即上游50 bp的特异性SNP会导致FT和FLC的表达水平发生改变, 进而影响3种甘蓝型油菜生态型开花时间的差异(Wu et al, 2019).Zou等(2014)利用DArT-Seq标记构建埃塞俄比亚芥双单倍群体的密集连锁图谱鉴定到24个与开花和出芽时间相关的QTL. ... A chromosome level genome assembly of a winter turnip rape (Brassica rapa L.) to explore the genetic basis of cold tolerance 1 2022 ... 随着21世纪初模式植物拟南芥(Arabidopsis thaliana)全基因组测序的完成(Theologis et al, 2000), 与其亲缘关系较近且同样具有重要经济价值的芸薹属植物逐渐成为植物学研究中重点关注的对象, 使得芸薹属植物的基因组测序工作获得了较为快速的发展.GenBank数据库显示, 自2011年首次完成并公开白菜基因组序列以来, 至今已有8个芸薹属物种完成了全基因组测序(表1).与此同时, 随着泛基因组(pan-genome)概念的提出(Tettelin et al, 2005), 研究者参考大豆、玉米和水稻等主要作物构建泛基因组的思路(Li et al, 2014; Lu et al, 2015; Zhao et al, 2018), 同步构建了包括甘蓝型油菜、白菜和冬油菜等芸薹属植物的泛基因组(Lin et al, 2014; Hurgobin et al, 2018; Song et al, 2020; Wu et al, 2022).总之, 这些全基因组与泛基因组图谱的构建为推动芸薹属植物的重测序研究, 包括利用基因组信息研究芸薹属植物的驯化起源、环境适应性以及重要性状形成的分子基础等方面的研究奠定了基础.本文将对近年来上述研究领域的新进展进行简要介绍. ... Di- and tri- but not monomethylation on histone H 3 lysine 36 marks active transcription of genes involved in flowering time regulation and other processes in Arabidopsis thaliana 1 2008 ... 此外, 研究者也对另一生物节律行为——抽薹开展了相关研究.植物的抽薹行为主要发生在花芽分化后, 也受开花调控的影响.通过重测序技术在两个大白菜早抽苔突变体ebm5-1和ebm5-2中鉴定到BrSDG8基因与大白菜抽薹相关, 并明确了该基因编码H3K4me3的蛋白质(Fu et al, 2020), 与H3K36me3共同参与拟南芥的FLC表达的激活(Pien et al, 2008; Xu et al, 2008).Wei等(2022)也成功挖掘出大白菜A07染色体上的3个延迟抽薹性状的潜在候选基因BraA07g029500、BraA07g029530和BraA07g030360.研究人员利用109个芥菜自然群体材料重测序数据揭示了CK2B1在芥菜茎膨大形成中的作用, 并进一步表明CK2B1与细胞周期G1/S转变因子E2Fa的相互作用, 阐明了控制芥菜茎膨大形成的机制, 为提高芥菜营养器官形成的产量提供了有效的分子靶点(Zhang et al, 2020). ... Resequencing 50 accessions of cultivated and wild rice yields markers for identifying agronomically important genes 1 2012 ... 目前, 全基因组重测序技术在动物、植物、微生物等研究中广泛使用, 在解析这些物种遗传分化和挖掘重要经济性状上发挥着重要作用(Kapur et al, 1995; Xu et al, 2012; Shang et al, 2020; Shi et al, 2020).芸薹属植物基因组的完成与公开, 为芸薹属植物的重测序研究提供了更多的可行性思路, 推动了芸薹属植物驯化起源、环境适应性以及重要性状基因挖掘等研究.因此, 本文总结分析了近年来芸薹属植物全基因组重测序研究结果, 围绕芸薹属植物驯化起源、环境适应性基因和重要性状基因3个方面进行介绍, 以期为日后更多的芸薹属植物研究提供思路与参考. ... Fine mapping and candidate gene analysis of the yellow petal gene ckpc in Chinese kale (Brassica oleracea L. var. alboglabra Bailey) by whole-genome resequencing 1 2019 ... 花色作为植物重要的性状, 不仅是昆虫授粉植物的主要视觉信号, 也是观赏性和景观美化的植物重要表型(Pearson, 1929; Zhang et al, 2018).同时, 对于评估杂交后的种子纯度也具有参考价值.因此, 鉴定花色性状的候选基因具有重要意义.以黄花芥菜和白色芥菜构建全基因重测序数据库分离鉴定到控制芥菜花色的基因BjPC1和BjPC2 (Zhang et al, 2018), 解析了芥菜花色受两个基因对的相互作用影响的分子基础, 弥补了此前研究的空白(Singh & Chauhan, 2011).在另一项研究中, 以黄花花椰菜和白花芥蓝为材料建立F2分离群体, 基于全基因组重测序明确了与类胡萝卜素积累相关的基因BoCCD4, 并发现产生这一现象的原因是CACAT转座子插入和In/Dels导致BoCCD4过早终止密码子, 为进一步了解甘蓝花色的遗传机制提供了依据(Yan et al, 2020).这一现象与此前通过图位克隆技术在黄花芥蓝中发现的变异相同(Xu et al, 2019).在其他物种的研究中, 发现BoCCD4的同源物的缺失会导致菊花花瓣由白色变为黄色(Ohmiya et al, 2006)、桃子产生黄色果肉(Falchi et al, 2013)和甘蓝型油菜产生黄色花色(Zhang et al, 2015). ... 芸薹属作物雄性不育研究进展 1 2007 ... 雄性不育是芸薹属作物中产生杂种优势的重要原因(许忠民等, 2007), 挖掘雄性不育性状相关的基因不仅对解析其遗传机制提供分子依据, 也可为芸薹属杂交育种提供重要的技术支撑.利用全基因组重测序分别在甘蓝型油菜突变体和大白菜突变体中鉴定到参与雄性和雌性不育调控的重要基因BnaC03g56870D (Teng et al, 2017)和Bra010198 (Tan et al, 2019), 为解析不同芸薹属植物雄性不育的遗传机制提供了重要依据.与此同时, 大白菜细胞质雄性不育(cytoplasmic male sterility, CMS)恢复系的In/Del标记SC718的发现, 为分子标记辅助育种培育和筛选新细胞质雄性不育恢复系提供了参考(Zhang et al, 2017).根据雄性不育和雄性不育恢复的关联, 利用细胞质雄性不育的甘蓝型油菜(B. napus)恢复系又解析了恢复基因Rfh, 为研究恢复基因和CMS基因之间的协同进化以及通过分子育种培育优良杂种提供了新的依据(Wei et al, 2019).此外, 研究者基于5个育种恢复系芥菜的全基因组重测序数据开发了KASPar标记, 用于辅助标记Rfo的转移和生产批次杂交水平测试(Gudi et al, 2020). ... 芸薹属作物雄性不育研究进展 1 2007 ... 雄性不育是芸薹属作物中产生杂种优势的重要原因(许忠民等, 2007), 挖掘雄性不育性状相关的基因不仅对解析其遗传机制提供分子依据, 也可为芸薹属杂交育种提供重要的技术支撑.利用全基因组重测序分别在甘蓝型油菜突变体和大白菜突变体中鉴定到参与雄性和雌性不育调控的重要基因BnaC03g56870D (Teng et al, 2017)和Bra010198 (Tan et al, 2019), 为解析不同芸薹属植物雄性不育的遗传机制提供了重要依据.与此同时, 大白菜细胞质雄性不育(cytoplasmic male sterility, CMS)恢复系的In/Del标记SC718的发现, 为分子标记辅助育种培育和筛选新细胞质雄性不育恢复系提供了参考(Zhang et al, 2017).根据雄性不育和雄性不育恢复的关联, 利用细胞质雄性不育的甘蓝型油菜(B. napus)恢复系又解析了恢复基因Rfh, 为研究恢复基因和CMS基因之间的协同进化以及通过分子育种培育优良杂种提供了新的依据(Wei et al, 2019).此外, 研究者基于5个育种恢复系芥菜的全基因组重测序数据开发了KASPar标记, 用于辅助标记Rfo的转移和生产批次杂交水平测试(Gudi et al, 2020). ... Rapid identification of yellow-flowered gene Bofc in cauliflower (Brassica oleracea var. botrytis) by bulked segregant analysis and whole-genome resequencing 1 2020 ... 花色作为植物重要的性状, 不仅是昆虫授粉植物的主要视觉信号, 也是观赏性和景观美化的植物重要表型(Pearson, 1929; Zhang et al, 2018).同时, 对于评估杂交后的种子纯度也具有参考价值.因此, 鉴定花色性状的候选基因具有重要意义.以黄花芥菜和白色芥菜构建全基因重测序数据库分离鉴定到控制芥菜花色的基因BjPC1和BjPC2 (Zhang et al, 2018), 解析了芥菜花色受两个基因对的相互作用影响的分子基础, 弥补了此前研究的空白(Singh & Chauhan, 2011).在另一项研究中, 以黄花花椰菜和白花芥蓝为材料建立F2分离群体, 基于全基因组重测序明确了与类胡萝卜素积累相关的基因BoCCD4, 并发现产生这一现象的原因是CACAT转座子插入和In/Dels导致BoCCD4过早终止密码子, 为进一步了解甘蓝花色的遗传机制提供了依据(Yan et al, 2020).这一现象与此前通过图位克隆技术在黄花芥蓝中发现的变异相同(Xu et al, 2019).在其他物种的研究中, 发现BoCCD4的同源物的缺失会导致菊花花瓣由白色变为黄色(Ohmiya et al, 2006)、桃子产生黄色果肉(Falchi et al, 2013)和甘蓝型油菜产生黄色花色(Zhang et al, 2015). ... 高通量测序技术及其在农业科学研究中的应用 1 2012 ... 全基因组重测序(whole-genome resequencing)通过对已有参考基因组序列的物种进行个体或群体全基因组测序, 从而检测出全基因组范围的单核苷酸多态性(single nucleotide polymorphism, SNP)、基因组结构变异(structure variation, SV)、插入缺失变异位点(insertion/deletion, In/Del)、拷贝数变异(copy number variation, CNV)等变异信息, 获得个体或群体分子遗传特征, 并进行重要经济性状基因筛选预测及遗传进化分析(李国治和邓卫东, 2018).自1977年利用第一代Sanger测序法(双脱氧末端终止法)绘制完整的基因图谱以来(Sanger et al, 1977), 目前已发展到第三代测序技术.第二代测序技术因具有高通量、低成本和高准确性等特点, 是目前全基因组重测序的主要测序手段, 主要包括454测序技术(Margulies et al, 2005)、Illumina/Solexa测序技术(Seo et al, 2005)和SOLiD测序技术(Mardis, 2008).454测序技术利用焦磷酸法测序, 使用串联平行合成原理, 通过将DNA片段连接在微球上, 同时在DNA合成过程中检测释放的光信号, 从而实现高通量的DNA测序; 其测序读长通常在400-700 bp之间, 测序误差率通常在1%-2%左右.Illumina/Solexa测序技术利用桥式PCR和碱基扩增原理, 通过反复周期性地加入荧光标记的核苷酸并检测释放的光信号, 实现高通量、高精度的DNA测序; 其测序读长通常可达到数百个碱基对, 测序通量是第一代测序技术的数千倍, 而测序错误率通常在0.1%-1%之间.SOLiD测序技术采用特殊的二碱基编码技术, 通过短DNA片段的反复循环合成和测序来实现高通量的DNA测序; 其测序读长通常为50-75个碱基对, 错误率通常在1%-2%左右.此外, 以Heliscope单分子技术、SMRT技术和纳米孔单分子技术为代表的第三代测序技术也逐步应用于科学研究.Heliscope技术和SMRT技术是利用荧光信号进行测序, 而纳米孔单分子技术则是利用不同碱基产生的电信号进行测序.这些三代测序技术的测序读长通常在数千个碱基对到数十万个碱基对之间, 但错误率通常在5%-15%之间.虽然第三代测序技术的长读长为研究提供了许多优势, 但其较低的精确度可能需要在数据分析中采取更加谨慎的措施, 以确保得到准确的测序结果(闫绍鹏等, 2012).随着测序技术的发展和改进, 高保真测序技术为科学研究带来了更多的机会.高保真测序技术利用荧光信号进行测序, 并通过多次循环合成和错误校正序列获取; 其测序读长可达数万个碱基对以上, 并且准确度可达99%以上(Wenger et al, 2019).这为解决基因组组装、基因变异检测等科学问题提供了更便捷的技术平台. ... 高通量测序技术及其在农业科学研究中的应用 1 2012 ... 全基因组重测序(whole-genome resequencing)通过对已有参考基因组序列的物种进行个体或群体全基因组测序, 从而检测出全基因组范围的单核苷酸多态性(single nucleotide polymorphism, SNP)、基因组结构变异(structure variation, SV)、插入缺失变异位点(insertion/deletion, In/Del)、拷贝数变异(copy number variation, CNV)等变异信息, 获得个体或群体分子遗传特征, 并进行重要经济性状基因筛选预测及遗传进化分析(李国治和邓卫东, 2018).自1977年利用第一代Sanger测序法(双脱氧末端终止法)绘制完整的基因图谱以来(Sanger et al, 1977), 目前已发展到第三代测序技术.第二代测序技术因具有高通量、低成本和高准确性等特点, 是目前全基因组重测序的主要测序手段, 主要包括454测序技术(Margulies et al, 2005)、Illumina/Solexa测序技术(Seo et al, 2005)和SOLiD测序技术(Mardis, 2008).454测序技术利用焦磷酸法测序, 使用串联平行合成原理, 通过将DNA片段连接在微球上, 同时在DNA合成过程中检测释放的光信号, 从而实现高通量的DNA测序; 其测序读长通常在400-700 bp之间, 测序误差率通常在1%-2%左右.Illumina/Solexa测序技术利用桥式PCR和碱基扩增原理, 通过反复周期性地加入荧光标记的核苷酸并检测释放的光信号, 实现高通量、高精度的DNA测序; 其测序读长通常可达到数百个碱基对, 测序通量是第一代测序技术的数千倍, 而测序错误率通常在0.1%-1%之间.SOLiD测序技术采用特殊的二碱基编码技术, 通过短DNA片段的反复循环合成和测序来实现高通量的DNA测序; 其测序读长通常为50-75个碱基对, 错误率通常在1%-2%左右.此外, 以Heliscope单分子技术、SMRT技术和纳米孔单分子技术为代表的第三代测序技术也逐步应用于科学研究.Heliscope技术和SMRT技术是利用荧光信号进行测序, 而纳米孔单分子技术则是利用不同碱基产生的电信号进行测序.这些三代测序技术的测序读长通常在数千个碱基对到数十万个碱基对之间, 但错误率通常在5%-15%之间.虽然第三代测序技术的长读长为研究提供了许多优势, 但其较低的精确度可能需要在数据分析中采取更加谨慎的措施, 以确保得到准确的测序结果(闫绍鹏等, 2012).随着测序技术的发展和改进, 高保真测序技术为科学研究带来了更多的机会.高保真测序技术利用荧光信号进行测序, 并通过多次循环合成和错误校正序列获取; 其测序读长可达数万个碱基对以上, 并且准确度可达99%以上(Wenger et al, 2019).这为解决基因组组装、基因变异检测等科学问题提供了更便捷的技术平台. ... Refinement of four major QTL for oil content in Brassica napus by integration of genome resequencing and transcriptomics 1 2022 ... 利用全基因组重测序挖掘芸薹属农作物中的重要农艺性状相关基因也是近年来研究热点之一.Yu等(2013)对150个结穗大白菜(B. rapa ssp. pekinensis cv. ‘Bre’)与不结穗大白菜(B. rapa ssp. chinensis cv. ‘Wut’)的杂交自交重组系群体进行全基因组重测序, 鉴定到18个与白菜头部性状相关的QTL和3个候选基因, 为解析白菜叶头及其他复杂性状的遗传基础研究提供了参考.Zhang B等(2021)对羽衣甘蓝(B. oleracea var. acephala)的F2和BC1群体测序表明其叶状性状是由BoLl-1的显性基因控制, 基于亲本等位基因分析表明叶状观赏羽衣甘蓝的BoLl-1基因的启动子区域内存在1个SNP和2个In/Del, 并发现该分子标记仅存在于小叶观赏甘蓝自交系中.Su等(2015)对卷心菜裂头易感自交系79-156与抗性自交系96-100杂交后群体进行重测序, 共定位到9个与抗裂头性状相关的QTL, 可解释39.4%-59.1%的表型变异, 另外还发现了3个影响相对较小的QTL (Hsr 3.2、4.2、9.2), 证明了卷心菜耐裂性的复杂遗传机制, 为提升卷心菜品质和产量提供了理论基础.Dong等(2021)在株高表型显著差异的甘蓝型油菜中鉴定到了2个与株高相关的QTL和影响株高的主效基因qPHA10.Yan等(2022)在油菜籽KenC-8 × N53-2 (KN DH)定位群体中检测到分别位于A08、A09、C03和C06染色体上的4个与含油量相关的QTL和297个与出油量相关的基因; Lu等(2019)在甘蓝型油菜中检测到了9个与含油量QTL重叠的基因, 其编码蛋白主要参与三酰甘油生物合成、脂肪酸合成和延伸.Zhao CJ等(2022)以甘蓝型油菜和白芥(Sinapis alba)为材料收集到204份重组自交系(RILs)的自然群体, 通过全基因组关联分析发现了一个新的含油量相关的主效QTL位点qA07.SOC和两个候选基因NRT1和BnaA07g12880D.Chai等(2021)基于油菜异常叶片132000B-3和正常叶片827-3杂交后代F2V3群体重测序数据, 在异常叶片形态油菜中筛选到影响油菜叶片形状和生长特性的候选基因BnA10g0422620和BnA10g0422610.Yang等(2021)基于183份不同用途的芥菜的重测序数据关联到与糖鞘脂(glycosphing-olipid, GSLs)含量相关的基因HAG1, 研究表明该基因的同源物MYB28是控制GSLs生物合成的主要转录因子(Hirai et al, 2007).Meng等(2023)对由芥菜毛状叶片密集的YJ27和无毛叶片的03B0307为亲本构建的F2群体使用重测序技术证明了bjuvb02g54610是芥菜毛状体发育的关键基因, 为芥菜毛状体形成和潜在靶基因的培育提供了重要信息. ... The genome sequence of allopolyploid Brassica juncea and analysis of differential homoeolog gene expression influencing selection 3 2016 ... Whole-genome sequencing of Brassica
本文的其它图/表
|

{kind=link}